R-TF-015-006 Clinical Investigation Report
Research Title
Clinical validation of an artificial intelligence-based medical device for the appropriateness of dermatology referrals from primary care.
Product Identification
| Information | |
|---|---|
| Device name | Legit.Health Plus (hereinafter, the device) |
| Model and type | NA |
| Version | 1.1.0.0 |
| Basic UDI-DI | 8437025550LegitCADx6X |
| Certificate number (if available) | MDR 000000 (Pending) |
| EMDN code(s) | Z12040192 (General medicine diagnosis and monitoring instruments - Medical device software) |
| GMDN code | 65975 |
| EU MDR 2017/745 | Class IIb |
| EU MDR Classification rule | Rule 11 |
| Novel product (True/False) | TRUE |
| Novel related clinical procedure (True/False) | TRUE |
| SRN | ES-MF-000025345 |
Throughout this document, references to "the device" refer to the investigational product identified above.
Device version under investigation and bridging to the CE-marked release
The investigational product evaluated in this report is the device at release version 1.1.0.0. The clinical-investigation window (2022-11-23 to 2025-05-06) spans development activities under the manufacturer's change-control process; the v1.1.0.0 release represents the frozen snapshot reported in this document. Device outputs used in the analyses reported here were generated on the v1.1.0.0 artefact under configuration control. The identity bridge between the investigational version and the CE-marked v1.1.0.0 release is held within the technical file and signed off by the Person Responsible for Regulatory Compliance (PRRC) under the manufacturer's configuration-management procedure.
Sponsor Identification and Contact
| Manufacturer data | |
|---|---|
| Legal manufacturer name | AI Labs Group S.L. |
| Address | Street Gran Vía 1, BAT Tower, 48001, Bilbao, Bizkaia (Spain) |
| SRN | ES-MF-000025345 |
| Person responsible for regulatory compliance | Alfonso Medela, Saray Ugidos |
| office@legit.health | |
| Phone | +34 638127476 |
| Trademark | Legit.Health |
| Authorized Representative | Not applicable (manufacturer is based in EU) |
Identification of the Clinical Investigation Plan (CIP)
| CIP | |
|---|---|
| Title of the clinical investigation | Pilot study for the clinical validation of an artificial intelligence algorithm to optimize the appropriateness of dermatology referrals. |
| Device under investigation | Legit.Health Plus |
| Protocol version | Version 1.0 |
| Date | 2022-04-07 |
| Protocol code | LEGIT.HEALTH_DAO_Derivación_O_2022 |
| Sponsor | AI Labs Group S.L. |
| Coordinating Investigator | Dr. Jesús Gardeazabal García and Dr. Rosa Mª Izu Belloso |
| Principal Investigator(s) | Dr. Jesús Gardeazabal García and Dr. Rosa Mª Izu Belloso |
| Investigational site(s) | Health Centre Sodupe-Güeñes, Health Centre Balmaseda, Health Centre Buruaga, and Health Centre Zurbaran |
| Ethics Committee | Comité de Ética de la Investigación con Medicamentos de Euskadi |
Trial Registrations
- ClinicalTrials.gov (NCT): NCT06228014
- EMA RWD Catalogue (EUPAS): EUPAS108167
Trial Registration and Data Availability
The investigation is registered on ClinicalTrials.gov under identifier NCT06228014 and in the EMA Real World Data Catalogue under identifier EUPAS108167. A public-facing results summary is available through those registrations. Patient-level data are not publicly accessible under the applicable patient-confidentiality and data-protection obligations (Regulation (EU) 2016/679 and Ley Orgánica 3/2018); de-identified datasets are available to competent authorities and to the notified body on formal request.
Research Team
- Osakidetza
- Dr. Jesús Gardeazabal García (Hospital Universitario Cruces)
- Dr. Rosa Mª Izu Belloso (Hospital Universitario Basurto)
Technical Support (Manufacturer)
- Mr. Alfonso Medela — Chief Technology Officer
- Mr. Taig Mac Carthy — Chief Executive Officer
Compliance Statement
The clinical investigation will be conducted according to the Clinical Investigation Plan (CIP) and other applicable guidances and regulations. This includes compliance with:
- The ethical principles originating from the
World Medical Association's Declaration of Helsinki - Harmonized standard
UNE-EN ISO 14155:2020 Regulation (EU) 2017/745 on medical devices (MDR), including the applicableGeneral Safety and Performance Requirements (GSPR)as outlined in Annex I, and the requirements ofAnnex XV(Chapter I and Chapter II, Section 3)- Harmonized standard
UNE-EN ISO 13485:2016 MDCG 2024-3for its structural and content expectations,MDCG 2021-8concerning application requirements, andMDCG 2020-10/1 Rev 1for safety reporting timelines and definitionsRegulation (EU) 2016/679(GDPR)- Spanish
Organic Law 3/2018on the Protection of Personal Data and guarantee of digital rights.
All data processing within the device is carried out in accordance with the highest standards of data protection and privacy. Patient information is managed in an encrypted manner to ensure confidentiality and security.
The research team assumes the role of Data Controller, responsible for the collection and management of study data. Legit.Health acts as the Data Processor and is not involved in the processing of patient data.
The storage and transfer of data comply with European data protection regulations. At the conclusion of the study, all information stored in the device will be permanently and securely deleted.
The device employs robust technical and organizational security measures to safeguard personal data against unauthorized access, alteration, loss, or processing.
Report Date
May 22, 2025.
The dates of versions, along with the version ID and the signature for the approval process for this document, can be found in the verified commits at the repository. This information is saved alongside the digital signature, to ensure the integrity of the document.
Report Author(s)
The full name, the ID and the signature for the authorship, as well as the approval process of this document, can be found in the verified commits at the repository. This information is saved alongside the digital signature, to ensure the integrity of the document.
Table of contents
Table of contents
- Research Title
- Product Identification
- Sponsor Identification and Contact
- Identification of the Clinical Investigation Plan (CIP)
- Trial Registration and Data Availability
- Research Team
- Compliance Statement
- Report Date
- Report Author(s)
- Table of contents
- Abbreviations and Definitions
- Summary
- Introduction
- Materials and methods
- Results
- Initiation and Completion Date
- Subject and Investigational Product Management
- Subject Demographics
- Clinical Investigation Plan (CIP) Compliance
- Protocol Deviations
- Analysis
- Referral adequacy
- Malignancy detection (secondary, exploratory)
- Impact of image quality (exploratory)
- Economic impact (secondary, supporting)
- Waiting list (secondary, supporting)
- Adverse Events and Adverse Reactions to the Product
- Product Deficiencies
- Subgroup Analysis for Special Populations
- Accounting for All Subjects
- Discussion and Overall Conclusions
- Ethical Aspects of Clinical Research
- Investigators and Administrative Structure of Clinical Research
- Report Annexes
Abbreviations and Definitions
- AE: Adverse Event
- AEMPS: Spanish Agency of Medicines and Medical Devices
- AEP: Adverse Reaction to Product
- AUC: Area Under the ROC Curve
- CAD: Computer-Aided Diagnosis
- CMD: Data Monitoring Committee
- CIP: Clinical Investigation Plan
- CUS: Clinical Utility Questionnaire
- DLQI: Dermatology Quality of Life Index
- GCP: Standards of Good Clinical Practice
- ICH: International Conference of Harmonization
- IFU: Instructions For Use
- IRB: Institutional Review Board
- N/A: Not Applicable
- NCA: National Competent Authority
- PI: Principal Investigator
- PPV: Positive Predictive Value
- NPV: Negative Predictive Value
- SAE: Serious Adverse Events
- SAEP: Serious Adverse Event to Product
- SUAEP: Serious and Unexpected Adverse Event to the Product
- SUS: System Usability Scale
Summary
This prospective observational analytical study of a longitudinal clinical case series evaluates whether the device can effectively improve the appropriateness of referrals from primary care to dermatology.
The investigation involves four primary care centres: Centro de Salud Sodupe-Güeñes, Centro de Salud Balmaseda, Centro de Salud Buruaga, and Centro de Salud Zurbaran. These centres refer patients to the Cruces and Basurto University Hospitals. A total of 127 patients were recruited, resulting in 198 dermatological images; the final analysis comprised 117 patients and 184 images after exclusion of cases lacking diagnostic confirmation.
Nature and positioning of the evidence
This investigation is a non-interventional observational clinical investigation of a CE-marked medical device, conducted under MDR Article 82 and the Spanish biomedical-research framework (Ley 14/2007 and Real Decreto 1090/2015 applicable at the time of conduct). Under MDCG 2020-6 Appendix III the evidence is ranked 2–4 for the device-versus-reference-standard diagnostic-accuracy analysis; per MDCG 2020-1 §4.4 it contributes Pillar 3 Clinical Performance evidence — measuring the clinician's referral decision when informed by the device's output on the Top-5 prioritised differential and malignancy gauge.
Introduction
Skin-related conditions present a significant challenge within primary care settings, frequently resulting in inconsistent diagnoses when compared to the evaluations conducted by dermatologists. This issue is made worse by a notable scarcity of dermatology specialists, particularly in less populous regions. This lack of specialists compels primary care practitioners to undertake dermatological evaluations, a field in which they may not have extensive expertise. Furthermore, the reliance on patient self-reporting during the diagnostic process can introduce a level of bias, potentially leading to inaccurate assessments.
To address these issues, Computer Aided Diagnosis (CAD) systems, including those using artificial intelligence, offer promising solutions for image interpretation and classification. Indeed, the purpose of this study is to clinically validate a device that increases the adequacy and efficiency of the referral process from primary care to dermatology and helps primary care practitioners perform dermatological assessments and triage.
By enhancing the accuracy and consistency of skin disease evaluations, the device can:
- Significantly improve the referral process
- Ensure patients are directed to specialist care when necessary
- Close the gap between primary care and specialized dermatology services
- Lead to better patient outcomes
Objectives
Primary Objective
To validate that the device is fit-for-purpose for optimising the adequacy of dermatology referrals.
Secondary Objectives
This study also validates that the device helps to:
- Reduce healthcare costs in secondary care through fewer unnecessary specialist visits
- Reduce dermatology waiting lists by optimizing referral processes
- Optimize clinical flows in Osakidetza, a northern Spanish public health service serving the Basque Country population
Acceptance Criteria
- AUC (area under the ROC curve) equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- specificity equal to or greater than 84.00% detecting malignancy.(User Group: Primary care practitioners)
- PPV (positive predictive value) equal to or greater than 40.00% detecting malignancy.(User Group: Primary care practitioners)
- NPV (negative predictive value) equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- equal to or greater than 15.00%.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 72.93%.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 45.00%.(User Group: Primary care practitioners)
- specificity equal to or greater than 66.11%.(User Group: Primary care practitioners)
- specificity equal to or greater than 47.00%.(User Group: Primary care practitioners)
- adequacy of referrals during in-person care equal to or greater than 50.00%.(User Group: Primary care practitioners)
- adequacy of referrals during in-person care equal to or greater than 38.90%.(User Group: Primary care practitioners)
- adequacy of referrals during remote care equal to or greater than 0.00%.(User Group: Primary care practitioners)
- adequacy of referrals during remote care equal to or greater than 67.00%.(User Group: Primary care practitioners)
- reduction in the number of days greater than 78.10%.(User Group: Primary care practitioners)
- reduction in the number of days equal to or greater than 30.00%.(User Group: Primary care practitioners)
- reduction in the number of days lower than 10.35.(User Group: Primary care practitioners)
Population
Adult patients (≥ 18 years) with skin conditions assessed in the primary care service of health centres referring to Cruces and Basurto University Hospitals.
Sample size
Considering a concordance rate of 55% between primary care and dermatology for referred lesions, a consensus agreed that a 15% reduction in inappropriate referrals would represent the minimum clinically important difference (MCID). To detect a 15% reduction in inappropriate referrals with 80% power at a two-sided 5% significance level under these assumptions, a target sample of 400 referred lesions from approximately 380 patients (some patients contributing more than one lesion) was pre-specified.
The pre-specified recruitment target was not achieved. The investigation closed with 127 patients and 198 images; the final analytical sample comprised 117 patients and 184 images after exclusion of 10 patients for lack of diagnostic confirmation. The resulting analytical sample is 49.5% of the pre-specified patient target. Reasons for the shortfall are documented in §Protocol Deviations. The primary endpoint observed in the analytical sample — a reduction in inappropriate referrals of 38% — comfortably exceeds the 15% MCID; the 95% confidence interval around the referral-rate difference excludes the null and supports the primary finding at the pre-specified α = 0.05 level within the analytical sample. The reduced sample does limit the precision of secondary and exploratory subgroup estimates; this limitation is honestly declared in §Limitations and is addressed by the PMCF confirmatory activity identified in R-TF-007-002.
Design and Methods
Design
This is a prospective observational analytical study of a longitudinal clinical case series.
Number of Subjects
127 patients were enrolled in the study, comprising 198 images of skin lesions.
The subjects were recruited from various health centres, with:
- Centro de Salud Buruaga: 74 patients
- Centro de Salud Balmaseda: 36 patients
- Centro de Salud Zurbaranbarri: 15 patients
- Centro de Salud Sodupe-Güeñes: 2 patients
Initiation Date
November 23, 2022.
Completion Date
May 6, 2025.
Duration
The duration of the study was 2 years, 5 months and 13 days, or 896 days including both the start and end dates. This period includes the recruitment, the specialist to review photos, and data analysis. The duration is 7.5 times longer than the initially estimated duration of 4 months, primarily due to challenges in patient recruitment. Specifically, at the Sodupe health centre, changes in the research team combined with high clinical workload hindered recruitment. At other health centres, significant care burden and overlapping vaccination campaigns substantially impacted the ability to recruit participants. Additionally, although some patients met inclusion criteria and were invited, some declined to consent to the use of their images in the study, further delaying image collection and preventing achievement of the expected sample size.
Methods
Each variable was characterised using frequency distributions for qualitative variables and central tendency statistics, such as the mean and median, along with variability statistics like the standard deviation (S.D.) or interquartile range for quantitative variables, in accordance with their distributional characteristics.
Sensitivity, specificity, positive and negative predictive values (PPV and NPV) and likelihood ratios (LR+ and LR-) were calculated by comparing both the results obtained using the device and those obtained with the referral criteria of primary care practitioners with the criteria used by specialists, considered the gold standard.
Confidence intervals (CIs) for sensitivity, specificity, PPV, NPV, and accuracy were estimated using the Wilson score method, which is recommended for proportions in diagnostic accuracy studies. For the AUC, CIs were computed using a bootstrap approach with 2000 iterations. Confidence intervals for LR+ and LR− were calculated using the log-transformation method based on the delta method, which accounts for the asymmetry and ratio nature of these estimates. All CIs were computed at a 95% confidence level.
Pre-specified acceptance criteria (for comparison against observed metrics)
The table below renders the pre-specified acceptance criteria carried by the Acceptance Criteria record for this investigation, shown alongside the observed values reported in §Analysis.
studyCode or folderSlug prop, or ensure this component is used within an Investigation document with a registered folder slug.Results
Within the analytical sample (117 patients / 184 images), the device reduced the number of inappropriate referrals by 38% compared with the primary care practitioner (from 81 to 50 inappropriate referrals across all modalities). The observed reduction exceeds the pre-specified 15% MCID for the primary endpoint.
The device also reduced the cumulative unnecessary waiting time observed in the analytical sample by 56% (from 929 days to 407 days across inappropriate referrals). The mean waiting time per referred patient in the analytical sample was reduced from 11.5 days to 5.0 days on the 56% assumption, and from 11.5 days to 7.1 days on the alternative 38% assumption (see §Waiting list for the derivation of both estimates); the conservative 7.1-day estimate is the primary figure cited in §Conclusions.
Comparison to prior validation of the device
The AUC for malignancy detection observed in this investigation (0.81) is lower than the AUC reported in earlier analytical-performance evaluations of the device (0.96). The present investigation is conducted on prospectively collected primary-care images across a broad mix of skin conditions in a real-world triage setting, which accounts for the difference in operating conditions compared with curated evaluations.
The device's referral-decision performance against the primary care practitioner in the analytical sample is summarised below. The threshold applied to the malignancy score to derive the device's binary referral decision for this comparison was 0.45; the origin of this threshold is declared in §Protocol Deviations.
| Metric | Device | Primary care practitioner | Difference |
|---|---|---|---|
| Sensitivity | 74.2%: 23/31 (95% CI: [56.8%-86.3%]) | 45.2%: 14/31 (95% CI: [29.2%-62.2%]) | +29 pp |
| Specificity | 67.3%: 103/153 (95% CI: [59.5%-74.2%]) | 47.1%: 72/153 (95% CI: [39.3%-54.9%]) | +20 pp |
Conclusions
Within the analytical sample, the device improves the appropriateness of primary-care dermatology referrals against the dermatologist reference standard. The observed 38% reduction in inappropriate referrals exceeds the pre-specified 15% MCID for the primary endpoint and the 95% CI around the referral-rate difference excludes the null at α = 0.05 within the analytical sample.
In the literature, primary care practitioners exhibit notably low sensitivity (approximately 45%) for identifying cases that require dermatology referral, implying a substantial risk that patients who genuinely need specialist care are not referred. In the present analytical sample the device achieved higher sensitivity and higher specificity than the primary care practitioner; this translated into a reduction in inappropriate referrals without increasing missed referrals within the sample.
Impact on waiting times
The cumulative unnecessary waiting time observed in the analytical sample was reduced by 56%. Expressed per referred patient, this corresponds to a reduction from a mean 11.5 days to approximately 7.1 days under the 38% inappropriate-referral-reduction assumption, and to approximately 5.0 days under the 56% cumulative-waiting-time-reduction assumption. The conservative 7.1-day estimate is reported as the headline figure; the 5.0-day estimate is reported as an upper bound. Both estimates are subject to the stated workforce-constant assumption.
The specificity of approximately 47% observed for the primary care practitioner in the analytical sample indicates a conservative referral pattern that may, despite appearing to contain costs, delay diagnosis for patients with time-critical skin cancers.
The device supports referral decision-making by reducing inappropriate referrals while preserving sensitivity for cases requiring specialist care within the analytical sample.
Clinical integration
The findings support evaluation of the device within teledermatology workflows in follow-on activities. Benefits to be confirmed under PMCF in larger, multi-region cohorts include optimised cost, expedited access for urgent cases, reduced pressure on dermatology services, and reduced delays caused by waiting-list congestion.
Case distribution
In the present analytical sample, 74% of cases were escalated to in-person consultations and 26% were resolved remotely, consistent with existing evidence that approximately 30% of primary-care dermatology referrals are of low complexity and could be managed at primary-care level when a validated triage tool is available.
Introduction
Skin-related diseases are a frequent reason for consultation in primary care1; some studies quantify it at approximately 5% of all consultations made, mainly by the working population. This is a considerable use of resources for health systems. For this reason, an efficient approach to referral and triage of cutaneous conditions is a key priority for many organisations.
Many studies show discrepancies in perspectives between the opinions of primary care practitioners and dermatologists, with percentages of agreement in their diagnoses ranging from 57%2 to 65.52%3 depending on the study. In general, primary care practitioners do not demonstrate adequate knowledge of skin diseases, their diagnosis and treatments4, partly due to the short training period in dermatology.
This limitation is also reflected in the effort and time required to estimate the degree of involvement of a patient or the stage of their condition. So much so, that it ends up being a very unrewarding task and can lead to poor adherence to the protocol and inadequate referrals.
Time consumption is of particular concern given that the number of medical professionals, especially in dermatology, is not sufficient for the current demand. Access of the general population to a dermatology specialist is complicated, due to the low number (3 dermatologists per 100,000 inhabitants)5, making it even more difficult in small population centres. Because of this, screening for dermatologic lesions must typically be performed by primary care practitioners, whose diagnostic capacity is even lower and can increase the risk of misdiagnosis.
In this regard, the literature shows a discordance of 55% to 65% between the primary care practitioner and the specialist3 and studies confirm several expected features: common dermatological diseases are often unrecognised or misdiagnosed by non-dermatologists, due to the particular profiles of common diagnoses in this activity (drug-induced rash, fungal infections)6.
In addition to these inherent limitations, one must also consider the risk of bias in cases where the preliminary examination is performed by the patient. This is especially true in cases where the patient knows that the treatment they receive will be determined by the information they provide. In addition, the medical team lacks the means to ensure that the values reported by the patient are true, which precludes external verification.
In recent years, there has been an increasing demand to develop Computer-Aided Diagnosis (CAD) systems that facilitate the detection of different conditions through algorithms. The device combines artificial intelligence and digital image processing to support the healthcare practitioner in interpreting the information contained in dermatological images. Advances in image recognition and artificial intelligence have led to innovations in the diagnosis of many conditions. It has been demonstrated that through artificial intelligence algorithms it is possible to classify photographs of lesions with a level of competence comparable to that of a medical expert7 8.
Indeed, artificial intelligence medical devices present a huge advance that not only brings reliability to the documentation process, but also allows greater precision when assessing a condition, triaging its urgency and measuring visual signs.
Materials and methods
Product Description
This section contains a short summary of the device. A complete description of the intended purpose, including device description, can be found in the record Legit.Health Plus description and specifications.
Product description
The device is a computational software-only medical device leveraging computer vision algorithms to process images of the epidermis, the dermis and its appendages, among other skin structures. Its principal function is to provide a wide range of clinical data from the analyzed images to assist healthcare practitioners in their clinical evaluations and allow healthcare provider organisations to gather data and improve their workflows.
The generated data is intended to aid healthcare practitioners and organizations in their clinical decision-making process, thus enhancing the efficiency and accuracy of care delivery.
The device should never be used to confirm a clinical diagnosis. On the contrary, its result is one element of the overall clinical assessment. Indeed, the device is designed to be used when a healthcare practitioner chooses to obtain additional information to consider a decision.
Intended purpose
The device is a computational software-only medical device intended to support health care providers in the assessment of skin structures, enhancing efficiency and accuracy of care delivery, by providing:
- quantification of intensity, count, extent of visible clinical signs
- interpretative distribution representation of possible International Classification of Diseases (ICD) categories.
Intended previous uses
No specific intended use was designated in prior stages of development.
Product changes during clinical research
The device maintained a consistent performance and features throughout the entire clinical research process. No alterations or modifications were made during this period.
Clinical Research Plan
This investigation includes a series of clinical cases to evaluate whether the device can effectively improve the appropriateness of referrals from primary care to dermatology.
The investigation also includes an analysis of cost reduction associated with the device, together with an analysis of waiting-list reduction and clinical-flow optimisation in Osakidetza.
The investigation is conducted at four primary care centres: Centro de Salud Sodupe-Güeñes, Centro de Salud Balmaseda, Centro de Salud Buruaga, and Centro de Salud Zurbaran. These centres refer patients to the Cruces and Basurto University Hospitals.
Objectives
The primary objective is to validate that the device is fit for purpose for optimising the appropriateness of dermatology referrals.
The secondary objectives are to validate that the device helps to reduce costs in secondary care, reduce dermatology waiting lists, and optimise clinical flow.
Acceptance Criteria
- AUC (area under the ROC curve) equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- specificity equal to or greater than 84.00% detecting malignancy.(User Group: Primary care practitioners)
- PPV (positive predictive value) equal to or greater than 40.00% detecting malignancy.(User Group: Primary care practitioners)
- NPV (negative predictive value) equal to or greater than 80.00% detecting malignancy.(User Group: Primary care practitioners)
- equal to or greater than 15.00%.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 72.93%.(User Group: Primary care practitioners)
- sensitivity equal to or greater than 45.00%.(User Group: Primary care practitioners)
- specificity equal to or greater than 66.11%.(User Group: Primary care practitioners)
- specificity equal to or greater than 47.00%.(User Group: Primary care practitioners)
- adequacy of referrals during in-person care equal to or greater than 50.00%.(User Group: Primary care practitioners)
- adequacy of referrals during in-person care equal to or greater than 38.90%.(User Group: Primary care practitioners)
- adequacy of referrals during remote care equal to or greater than 0.00%.(User Group: Primary care practitioners)
- adequacy of referrals during remote care equal to or greater than 67.00%.(User Group: Primary care practitioners)
- reduction in the number of days greater than 78.10%.(User Group: Primary care practitioners)
- reduction in the number of days equal to or greater than 30.00%.(User Group: Primary care practitioners)
- reduction in the number of days lower than 10.35.(User Group: Primary care practitioners)
Design
This is a prospective observational analytical study of a longitudinal clinical case series.
The investigation does not involve an active or control group, as it is focused on evaluation of the device against the dermatologist reference standard in a real-world primary-care setting. The device output is generated on photographs collected prospectively under the CIP; the primary care practitioner's referral decision is recorded at the point of consultation without device output being visible, and the device-versus-practitioner comparison is performed retrospectively against the dermatologist reference standard.
Ethical considerations
Ethics Committee Approval
Approved by CEIm of Euskadi on 2022-11-23, reference number PS2022074. A substantial modification (modificación relevante 1) was subsequently reviewed and approved by the same Ethics Committee; the scope of the modification is recorded in §Protocol Deviations. No subject was enrolled before the favourable opinion of 2022-11-23.
This study adhered to international Good Clinical Practice (GCP) guidelines, the Declaration of Helsinki in its latest amendment, and applicable international and national regulations. As applicable, approval from the relevant Ethics Committee was obtained prior to the initiation of the study. When applicable, modifications to the protocol were reviewed and approved by the Principal Investigator (PI) and subsequently evaluated by the Ethics Committee before subjects were enrolled under a modified protocol.
This study was conducted in compliance with European Regulation 2016/679, of 27 April, concerning the protection of natural persons with regard to the processing of personal data and the free movement of such data (General Data Protection Regulation, GDPR), and Organic Law 3/2018, of 5 December, on the Protection of Personal Data and the guarantee of digital rights. In accordance with these regulations, no data enabling the personal identification of participants was collected, and all information was managed securely in an encrypted format.
Participants were informed both orally and in writing about all relevant aspects of the study, with the information being tailored to their level of understanding. They were provided with a copy of the informed consent form and the accompanying patient information sheet. Adequate time was given to patients to ask questions and fully comprehend the details of the study before providing their consent.
The PI was responsible for the preparation of the informed consent form, ensuring it included all elements required by the International Conference on Harmonisation (ICH), adhered to current regulatory guidelines, and complied with the ethical principles of GCP and the Declaration of Helsinki.
The original signed informed consent forms were securely stored in a restricted access area under the custody of the PI. These documents remained at the research site at all times. Participants were provided with a copy of their signed consent form for their records.
Data confidentiality
Current legislation will be complied with in terms of data confidentiality protection (European Regulation 2016/679, of 27 April, on the protection of natural persons with regard to the processing of personal data and the free movement of such data and Organic Law 3/2018, of 5 December, on Personal Data Protection and guarantee of digital rights). For this purpose, when applicable, each participant will receive an alphanumeric identification code in the study that will not include any data allowing personal identification (coded CRD). The Principal Investigator will have an independent list that will allow the connection of the identification codes of the patients participating in the study with their clinical and personal data. This document will be filed in a secure area with restricted access, under the custody of the Principal Investigator and will never leave the centre.
Once the paper CRDs are completed and closed by the Principal Investigator, the data will be transferred to a database.
As in the CRDs, the Database will comply with current legislation in terms of data confidentiality protection (European Regulation 2016/679, of 27 April, on the protection of natural persons about the processing of personal data and the free movement of such data and Organic Law 3/2018, of 5 December, on the Protection of Personal Data and guarantee of digital rights) in which no data allowing personal identification of patients will be included.
Data Quality Assurance
The Principal Investigator is responsible for reviewing and approving the protocol, signing the Principal Investigator commitment, guaranteeing that the persons involved in the centre will respect the confidentiality of patient information and protect personal data, and reviewing and approving the final study report together with the sponsor. All the clinical members of the research team assess the eligibility of the patients in the study, inform and request written informed consent, collect the source data of the study in the clinical record and transfer them to the Data Collection Notebook (DCN) or Data Collection Forms (CRF).
Subject Population (inclusion/exclusion criteria and sample size)
The study enrolled patients who fulfilled the recruitment criteria. As a result, we recruited 127 patients from different health centres: Buruaga recruited 74 patients, Balmaseda 36 patients, Zurbaranbarri 15 patients, and Sodupe 2 patients.
Among all the 127 patients, we collected a total of 198 images using smartphones and dermatoscopes. Overall, patients with pigmented lesions tend to have more images on average because they include both clinical and dermatoscopic pictures.
Inclusion Criteria
- Patients with skin conditions.
- Patients aged 18 years or older.
- Patients who have signed the informed consent for the study.
Exclusion Criteria
- Patient who, at the investigator's discretion, will not comply with the study procedures.
Treatment
Patients participating in this study did not receive any specific treatment as part of the research protocol.
Concomitant Medication/Treatment
Patients continued their regular prescribed medications and treatments as directed by their primary healthcare providers. No additional medications or treatments were administered as part of this study.
Statistical Analysis
A total of 127 patients were enrolled. Ten patients were excluded due to lack of diagnostic confirmation; the final analytical sample therefore comprised 117 patients and 184 images.
The average patient age was 60 years, with a median age of 65 and a standard deviation of 20. Patient ages ranged from 19 to 98 years.
Among the 127 patients enrolled, 81 were female and 46 were male.
These patients, as diagnosed by the dermatologist, presented a range of dermatological conditions of which 14 images (from 14 patients) were malignant. The most common conditions were keratoses, melanocytic nevus, psoriasis, and basal cell carcinoma. A full list of the conditions with the number of occurrences is shown below. Conditions are associated with individual photographs, as a single patient may contribute more than one image corresponding to different pathologies:
| Condition | Count | Percentage |
|---|---|---|
| seborrheic keratosis | 33 | 17.93 |
| actinic keratosis | 27 | 14.67 |
| melanocytic nevus | 24 | 13.04 |
| psoriasis | 9 | 4.89 |
| basal cell carcinoma | 8 | 4.35 |
| keratoacanthoma | 5 | 2.72 |
| asteatotic eczema | 5 | 2.72 |
| bullous pemphigoid | 5 | 2.72 |
| plaque psoriasis | 4 | 2.17 |
| spider telangiectasis | 3 | 1.63 |
| guttate psoriasis | 3 | 1.63 |
| pyogenic granuloma | 3 | 1.63 |
| eczema | 3 | 1.63 |
| nummular dermatitis | 3 | 1.63 |
| amelanotic malignant melanoma | 3 | 1.63 |
| erythema | 3 | 1.63 |
| erythema multiforme | 2 | 1.09 |
| necrobiosis lipoidica | 2 | 1.09 |
| epidermoid cyst | 2 | 1.09 |
| malignant melanoma | 2 | 1.09 |
| intradermal nevus | 2 | 1.09 |
| lichenoid keratosis | 2 | 1.09 |
| dermatitis | 2 | 1.09 |
| atopic dermatitis | 2 | 1.09 |
| seborrheic dermatitis | 2 | 1.09 |
| alopecia | 2 | 1.09 |
| nail fragility | 2 | 1.09 |
| pigmented basal cell carcinoma | 1 | 0.54 |
| mixed epithelioid and spindle cell melanoma | 1 | 0.54 |
| dyshidrotic eczema | 1 | 0.54 |
| alopecia areata | 1 | 0.54 |
| common warts | 1 | 0.54 |
| folliculitis decalvans | 1 | 0.54 |
| telogen effluvium | 1 | 0.54 |
| lobular capillary haemangioma | 1 | 0.54 |
| anogenital warts | 1 | 0.54 |
| lentigo | 1 | 0.54 |
| fibroepithelial polyp | 1 | 0.54 |
| neurofibroma | 1 | 0.54 |
| melanoma in situ | 1 | 0.54 |
| trichilemmal cyst | 1 | 0.54 |
| folliculitis | 1 | 0.54 |
| dermatofibroma | 1 | 0.54 |
| focal palmoplantar keratoderma | 1 | 0.54 |
| zoster | 1 | 0.54 |
| lichen simplex chronicus | 1 | 0.54 |
| benign lymphocytic infiltration of the skin | 1 | 0.54 |
| actinic cheilitis | 1 | 0.54 |
Regarding the diagnostic recording of primary care practitioners, initial diagnoses were frequently captured using open-text formats that do not strictly follow a standardised nomenclature such as ICD, consistent with natural clinical documentation practices in primary care. For this investigation, all primary-care-practitioner diagnoses were subsequently standardised and harmonised for comparative analysis with the standardised dermatologist assessments. This harmonisation ensured that initial open-text diagnoses could be accurately mapped to standard dermatological terminology for comparative evaluation of referral appropriateness. Examples of initial open-text formats included "ca.basocelular drch", "granuloma/ca.basocelular", "ID basocelular", "pie izq" (anatomical location), and clinical observations such as "post inflamatorio".
Regarding the diagnostic recording of dermatologists, a relatively standardised terminology was used, although it did not strictly follow the ICD standard. Examples of dermatologist diagnoses include seborrheic keratosis, melanocytic nevi without atypia, basal cell carcinoma of the scalp and neck, and porokeratosis (cornoid lamella).
For five specific cases, the initial dermatologist diagnosis included two distinct conditions with differing levels of malignancy. To resolve these discrepancies and establish a definitive reference diagnosis, a second expert dermatologist was consulted and the final diagnosis was reached through consensus. Details are provided below:
| Photo ID | Referral | Initial dermatologist diagnosis | Second dermatologist's diagnosis |
|---|---|---|---|
| 032_01 | 0 | Inflammatory lesion vs Bowen's disease | Actinic keratosis |
| 034_01 | 0 | Tumor-like lesion / keratoacanthoma vs epidermoid cyst | Keratoacanthoma |
| 036_01 | 1 | Pigmented papular lesion on the vertex | Dermatofibroma |
| 046_01 | 0 | Actinic cheilitis vs tumoral lesion | Actinic cheilitis |
| 099_01 | 1 | Irritated seborrheic keratosis vs basal cell carcinoma and MM | Seborrheic keratosis |
Results
Initiation and Completion Date
The study started on November 23, 2022 and ended on May 6, 2025.
Subject and Investigational Product Management
The investigational product, a software medical device, was deployed and managed under the manufacturer's configuration-management and information-security procedures. Controls comprised version control of the device artefact, controlled access for authorised users via individual authenticated accounts, secure deployment environments, and systematic logging of usage. Device outputs for the analyses reported in this document were generated on the v1.1.0.0 artefact identified in §Product Identification and are reproducible on that artefact; the algorithm version identifier and timestamp of scoring are logged for each image under the sponsor's records-retention procedure. Accountability and traceability of the investigational software are ensured through audit trails, user authentication and documentation of updates throughout the investigation.
Subject Demographics
The average patient age was 60 years, with a median age of 65 and a standard deviation of 20. Patient ages ranged from 19 to 98 years.
Among the 127 patients recruited for the study, 81 were female and 46 were male.
All participants in this study were from Spain.
Gender: Men: 46 (36.2%), Women: 81 (63.8%)
| Age Group | Count | Percentage |
|---|---|---|
| Newborn (birth to 1 month) | 0 | 0.0% |
| 1 month to 2 years | 0 | 0.0% |
| 2 to 12 years | 0 | 0.0% |
| 12 to 21 years | 3 | 2.4% |
| Age >= 22 and < 65 | 58 | 45.7% |
| Age >= 65 | 66 | 52.0% |
| Fitzpatrick Phototype | Count | Percentage |
|---|---|---|
| Phototype I | 86 | 67.7% |
| Phototype II | 29 | 22.9% |
| Phototype III | 9 | 7.5% |
| Phototype IV | 2 | 1.5% |
| Phototype V | 1 | 0.5% |
| Phototype VI | 0 | 0.0% |
The analytical cohort is predominantly Fitzpatrick I–III (98.1%); Fitzpatrick V is represented by a single participant (0.5%) and Fitzpatrick VI is not represented. This investigation does not, on its own, support performance claims on Fitzpatrick V or VI populations; phototype coverage for darker skin tones is addressed by dedicated phototype-bridging evidence at Clinical Evaluation level (R-TF-015-003) and by the PMCF Plan (R-TF-007-002). All participants were recruited from primary care in the Basque Country (Spain); generalisability to other European healthcare systems, patient populations and primary-care workflows remains to be established via the PMCF activities.
Clinical Investigation Plan (CIP) Compliance
The investigation was conducted in accordance with the CIP subject to the deviations declared and described in §Protocol Deviations. Compliance with the CIP was monitored throughout the investigation to uphold the integrity and validity of the research findings. Each deviation is documented with its rationale and the associated regulatory handling.
Protocol Deviations
The following deviations from the CIP are declared for this investigation:
-
Recruitment shortfall (major protocol deviation). The pre-specified sample size of 400 referred lesions from approximately 380 patients was not achieved. The investigation closed with 127 patients / 198 images; the final analytical sample is 117 patients / 184 images (49.5% of the patient target). Contributing factors include clinical-workload challenges at participating primary-care centres (notably the Sodupe-Güeñes centre), changes in research-team composition during the investigation window, and overlapping vaccination-campaign obligations that reduced available recruitment slots. The primary endpoint was nonetheless met within the analytical sample, as the observed 38% reduction in inappropriate referrals comfortably exceeds the pre-specified 15% MCID; however, the reduced sample limits precision of secondary and exploratory estimates (in particular malignancy-detection subgroup estimates), and this limitation is declared in §Limitations. Independent-sample confirmation of the primary endpoint is committed to under the PMCF Plan (R-TF-007-002).
-
Duration extension (major protocol deviation). The pre-specified total duration was 4 months; the actual duration from study initiation to study closure was 2 years, 5 months and 13 days (896 days). The extension was driven by the recruitment factors listed under deviation 1 and was notified to the Ethics Committee under the substantial modification recorded at §Ethics Committee Approval.
-
Referral-decision threshold (methodological deviation). The CIP pre-specified that the referral-decision threshold applied to the device's malignancy output in the primary analysis would be the threshold documented in the IFU for referral output. The primary-analysis results reported in §Analysis additionally apply a threshold of 0.45 to the malignancy score to derive the device's binary referral decision for comparison against the primary care practitioner. This threshold was informed by a Youden-J threshold sweep on the analytical sample and is therefore not an independently pre-specified operating point. The risk of optimistic bias from in-sample threshold selection is declared in §Limitations. Independent-sample confirmation of the primary analysis at a pre-specified operating threshold is committed to under the PMCF Plan.
-
Post-hoc image-quality stratification (methodological deviation). Analyses stratified by DIQA score (cut-offs 5, 6 and 7) were not pre-specified in the CIP. These analyses are reported as exploratory / supporting in §Impact of image quality; they are not used for primary inference. A pre-specified operating-quality threshold for the device's referral output is committed to under the PMCF Plan.
-
Second-dermatologist adjudication on five discordant cases. For five specific cases the initial dermatologist diagnosis included two distinct conditions with differing levels of malignancy; a second expert dermatologist was consulted and the final reference diagnosis was established through consensus. The five adjudicated cases are tabulated in §Subject Population. The adjudication procedure is not a primary-analysis deviation but is declared here for completeness; the adjudicated diagnoses were treated as the reference standard for those five cases.
Analysis
Referral adequacy
Dermatologists managing teledermatology cases typically follow one of two pathways: review and resolve cases remotely, or schedule an in-person consultation. In the analytical sample, dermatologists opted for in-person consultations in 74% of cases and resolved 26% remotely. This aligns with existing literature, which suggests that approximately 30% of primary care dermatology referrals are of low complexity and could potentially be managed within primary care.
Among the 26% of cases addressed remotely, 3 patients (6%) ultimately required referral to a dermatologist. In this subgroup, primary care practitioners did not identify any of the necessary referrals — sensitivity 0.0%: 0/3 (95% CI: [0.0%-56.1%]) and specificity 66.7%: 30/45 (95% CI: [52.1%-78.6%]). In contrast, the device achieved a sensitivity of 33.3%: 1/3 (95% CI: [6.1%-79.2%]) at the same specificity, demonstrating capacity to detect referral-worthy cases that were otherwise missed, without increasing false positives. The very small number of true-positive cases in this subgroup (n = 3) is noted as a constraint on inference.
For patients who underwent in-person consultation, 28 cases (21%) were determined to require referral to dermatology. In this subset, the primary care practitioners achieved a sensitivity of 50.0%: 14/28 (95% CI: [32.6%-67.4%]) and specificity of 38.9%: 42/108 (95% CI: [30.2%-48.3%]), whereas the device achieved a sensitivity of 78.6%: 22/28 (95% CI: [60.5%-89.8%]) and specificity of 67.6%: 73/108 (95% CI: [58.3%-75.7%]).
When aggregating both teledermatology and in-person cases, using the referral-decision threshold of 0.45 declared in §Protocol Deviations, the device achieved a sensitivity of 74.2%: 23/31 (95% CI: [56.8%-86.3%]) and specificity of 67.3%: 103/153 (95% CI: [59.5%-74.2%]). By contrast, the primary care practitioner achieved a sensitivity of 45.2%: 14/31 (95% CI: [29.2%-62.2%]) and specificity of 47.1%: 72/153 (95% CI: [39.3%-54.9%]) on the same sample.
Within the analytical sample, the device identified referral-appropriate cases that were missed by the primary care practitioner — particularly important in scenarios where missed diagnoses can have serious consequences. In the malignancy-subset narrative, the device correctly flagged two cases of basal cell carcinoma (patients 88 and 58) and one case of amelanotic malignant melanoma (patient 23) that were missed by the primary care practitioner. Beyond malignancies, the device also flagged additional conditions requiring referral that were missed by the primary care practitioner, including keratoacanthoma (5 patients) and pyogenic granuloma (3 patients).
In summary, the results within the analytical sample point to two opportunities for improving diagnostic efficiency and patient safety:
- Teledermatology cases, where the device can support reduced missed referrals.
- In-person consultations, where the device can support a reduction in inappropriate referrals to dermatology.
Malignancy detection (secondary, exploratory)
Malignancy detection is reported here as a secondary, exploratory analysis. The number of malignant cases in the analytical sample (n = 14, including 2 melanoma, 1 amelanotic malignant melanoma, 1 melanoma in situ, 1 mixed epithelioid/spindle cell melanoma, 8 basal cell carcinoma and 1 pigmented basal cell carcinoma) is insufficient to support a confirmatory claim about malignancy-specific sensitivity; the estimates below are reported with their 95% CIs and are to be confirmed in a dedicated, powered study under the PMCF Plan.
The analytical sample included 184 images, of which 170 (92%) were benign and 14 (8%) malignant.
For the original images, the device achieved a sensitivity of 57.1%: 8/14 (95% CI: [32.6%-78.6%]), a specificity of 93.5%: 159/170 (95% CI: [88.8%-96.3%]), a positive predictive value (PPV) of 42.1%: 8/19 (95% CI: [23.1%-63.7%]), and a negative predictive value (NPV) of 96.4%: 159/165 (95% CI: [92.3%-98.3%]). The corresponding likelihood ratios were LR+ = 8.83 (95% CI: [5.48–14.24]) and LR− = 0.46 (95% CI: [0.27–0.77]).
Statistical interpretation of PPV in the context of class imbalance
The PPV of 42.1% reflects the low prevalence of malignant cases in the analytical sample (14 of 184 images). Low disease prevalence yields lower positive predictive values even at high specificity. The negative predictive value of 96.4% indicates that, within the analytical sample, images classified by the device as low-risk had a 96% probability of being benign at the applied operating threshold. The AUC of 0.815 and specificity of 93.5% indicate discriminatory ability within the sample, and LR+ of 8.83 indicates that a positive classification is approximately 8.8 times more likely in a truly malignant lesion than in a benign one.
The lower bound of the 95% CI for malignancy sensitivity (32.6%) is insufficient to support a screening or rule-out claim for malignancy on the basis of this investigation alone. A screening or rule-out claim requires confirmation in a dedicated, powered study; this confirmation is committed to under the PMCF Plan (R-TF-007-002).
Performance metrics for both original and cropped image inputs are summarised below:
| Sensitivity | Specificity | PPV | NPV | LR+ | LR- | |
|---|---|---|---|---|---|---|
| The device (original images) | 57.1%: 8/14 (95% CI: [32.6%-78.6%]) | 93.5%: 159/170 (95% CI: [88.8%-96.3%]) | 42.1%: 8/19 (95% CI: [23.1%-63.7%]) | 96.4%: 159/165 (95% CI: [92.3%-98.3%]) | 8.83 (95% CI: [5.48-14.24]) | 0.46 (95% CI: [0.27-0.77]) |
| The device (cropped images) | 42.9%: 6/14 (95% CI: [21.4%-67.4%]) | 92.9%: 158/170 (95% CI: [88.1%-95.9%]) | 33.3%: 6/18 (95% CI: [16.3%-56.3%]) | 95.2%: 158/166 (95% CI: [90.8%-97.5%]) | 6.07 (95% CI: [3.26-11.32]) | 0.61 (95% CI: [0.36-1.04]) |
To further characterise model performance, the continuous malignancy score was analysed across decision thresholds. The area under the ROC curve (AUC) on the analytical sample was 81.5% (95% CI: [66.6%-94.1%]), indicating discriminatory ability across thresholds in the sample. When images were cropped to focus more precisely on the lesion, the AUC was 0.82, consistent with improved performance for lesion-centred inputs.
A threshold sweep was performed on the analytical sample with Youden's J index reported as a summary measure at each cut-off (table below). This sweep is exploratory and does not establish a pre-specified operating threshold; the selection of any operating threshold for the device's referral output is subject to the PMCF confirmatory activity identified in the PMCF Plan.
| Malignancy threshold | Sensitivity | Specificity | NPV | PPV | Youden's J index |
|---|---|---|---|---|---|
| 0.0500 | 0.7857 | 0.6235 | 0.9725 | 0.1467 | 0.4092 |
| 0.1000 | 0.7143 | 0.7647 | 0.9701 | 0.2000 | 0.4790 |
| 0.1500 | 0.6429 | 0.7824 | 0.9638 | 0.1957 | 0.4252 |
| 0.2000 | 0.6429 | 0.8176 | 0.9653 | 0.2250 | 0.4605 |
| 0.2500 | 0.6429 | 0.8588 | 0.9669 | 0.2727 | 0.5017 |
| 0.3000 | 0.6429 | 0.8706 | 0.9673 | 0.2903 | 0.5134 |
| 0.3500 | 0.6429 | 0.8882 | 0.9679 | 0.3214 | 0.5311 |
| 0.4000 | 0.5714 | 0.8941 | 0.9620 | 0.3077 | 0.4655 |
| 0.4500 | 0.5714 | 0.9235 | 0.9632 | 0.3810 | 0.4950 |
| 0.5000 | 0.5714 | 0.9353 | 0.9636 | 0.4211 | 0.5067 |
| 0.5500 | 0.5714 | 0.9529 | 0.9643 | 0.5000 | 0.5244 |
| 0.6000 | 0.5000 | 0.9588 | 0.9588 | 0.5000 | 0.4588 |
| 0.6500 | 0.4286 | 0.9647 | 0.9535 | 0.5000 | 0.3933 |
| 0.7000 | 0.3571 | 0.9706 | 0.9483 | 0.5000 | 0.3277 |
| 0.7500 | 0.2857 | 0.9824 | 0.9435 | 0.5714 | 0.2681 |
| 0.8000 | 0.2143 | 0.9882 | 0.9385 | 0.6000 | 0.2025 |
| 0.8500 | 0.0000 | 1.0000 | 0.9239 | 0.0000 | |
| 0.9000 | 0.0000 | 1.0000 | 0.9239 | 0.0000 | |
| 0.9500 | 0.0000 | 1.0000 | 0.9239 | 0.0000 |
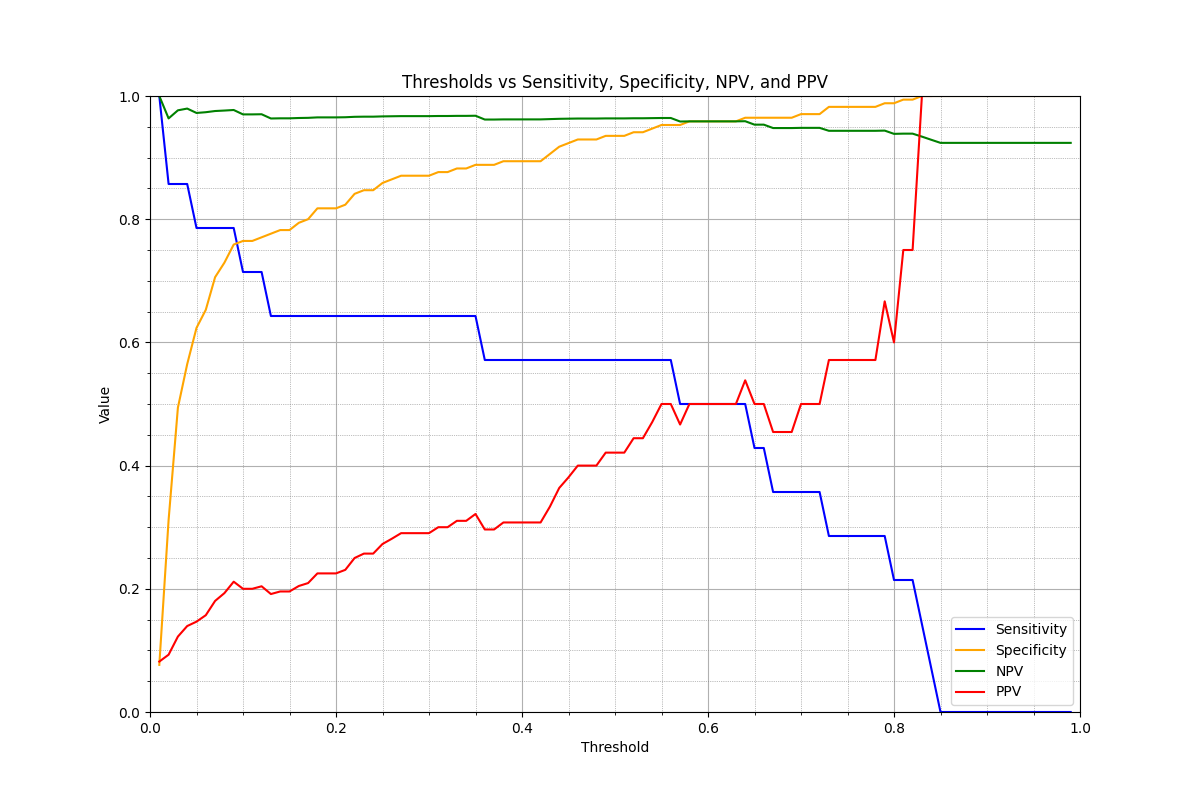
Figure 1. Sensitivity, specificity and Youden's J index across malignancy-score thresholds on the analytical sample (n = 184 images, 14 malignant).
Impact of image quality (exploratory)
The impact of image quality on device performance for malignancy detection and referral decision-making was examined as an exploratory analysis (not pre-specified in the CIP; see §Protocol Deviations). Image quality was assessed using the DIQA algorithm. Performance metrics — sensitivity, specificity, AUC, and the number of positive and negative cases — are reported at several DIQA thresholds below.
The first table shows device performance for malignancy detection at successive DIQA cut-offs. Sensitivity and AUC increase with higher DIQA thresholds while specificity remains relatively stable. The number of positive and negative cases changes as the sample is restricted to higher-quality images. At a DIQA cut-off of 7 the device reached sensitivity 100.0%: 2/2 (95% CI: [34.2%-100.0%]) and specificity 94.7%: 72/76 (95% CI: [87.2%-97.9%]); the very small number of positive cases (n = 2) limits inference and the result is reported as exploratory only.
| DIQA threshold | Sensitivity | Specificity | AUC |
|---|---|---|---|
| All | 57.1%: 8/14 (95% CI: [32.6%-78.6%]) | 93.5%: 159/170 (95% CI: [88.8%-96.3%]) | 81.5%, (95% CI: [66.6%-94.1%]) |
| 5 | 66.7%: 8/12 (95% CI: [39.1%-86.2%]) | 92.8%: 141/152 (95% CI: [87.5%-95.9%]) | 86.0%, (95% CI: [71.6%-97.1%]) |
| 6 | 62.5%: 5/8 (95% CI: [30.6%-86.3%]) | 94.8%: 110/116 (95% CI: [89.2%-97.6%]) | 86.0%, (95% CI: [65.2%-99.4%]) |
| 7 | 100.0%: 2/2 (95% CI: [34.2%-100.0%]) | 94.7%: 72/76 (95% CI: [87.2%-97.9%]) | 99.3%, (95% CI: [96.1%-100.0%]) |
The second table presents the same stratification for the referral-decision analysis. Sensitivity increases with higher image quality while specificity decreases. AUC improves with higher DIQA scores, consistent with the expected influence of image quality on device output. A pre-specified operating DIQA threshold for the referral output is committed to under the PMCF Plan.
| DIQA threshold | Sensitivity | Specificity | AUC |
|---|---|---|---|
| All | 74.2%: 23/31 (95% CI: [56.8%-86.3%]) | 67.3%: 103/153 (95% CI: [59.5%-74.2%]) | 74.3%, (95% CI: [63.6%-83.9%]) |
| 5 | 75.0%: 21/28 (95% CI: [56.6%-87.3%]) | 66.2%: 90/136 (95% CI: [57.9%-73.6%]) | 75.7%, (95% CI: [64.6%-86.1%]) |
| 6 | 76.2%: 16/21 (95% CI: [54.9%-89.4%]) | 66.0%: 68/103 (95% CI: [56.4%-74.4%]) | 74.7%, (95% CI: [61.9%-86.3%]) |
| 7 | 80.0%: 8/10 (95% CI: [49.0%-94.3%]) | 63.2%: 43/68 (95% CI: [51.4%-73.7%]) | 77.5%, (95% CI: [63.2%-89.9%]) |
Economic impact (secondary, supporting)
One of the secondary objectives of this investigation was to evaluate the potential economic benefit of integrating the device into the referral process, specifically by reducing inappropriate specialist consultations and the associated waiting times in secondary care.
A key metric for economic efficiency is the unnecessary waiting time, defined as the total waiting days incurred by patients referred to secondary care but ultimately not diagnosed with conditions requiring such referral. Reducing inappropriate referrals has a direct impact on healthcare costs and resource optimisation.
Overall referral population
In the current primary-care pathway across all modalities within the analytical sample, 81 cases were deemed inappropriate referrals, resulting in a cumulative waiting time of 929 days. Using the device with the referral-decision threshold of 0.45 declared in §Protocol Deviations, the corresponding figures were 50 inappropriate referrals and 407 cumulative waiting days. These observations correspond to a 38% reduction in inappropriate referrals and a 56% reduction in cumulative waiting time within the analytical sample.
No annualised monetary extrapolation is made on the basis of this analytical sample alone. Cost-saving extrapolations to the population served by Osakidetza, or to other European healthcare systems, require assumptions (per-consultation unit cost, referral-volume denominator, workforce assumptions) that are not derivable from the analytical sample of 117 patients and that are subject to the generalisability caveats declared in §Limitations. The reduction in inappropriate referrals observed in the analytical sample provides a basis for PMCF activities that will quantify cost-impact under documented assumptions; this is identified in the PMCF Plan.
Teledermatology subgroup
In the subset of cases managed through teledermatology, primary care practitioners generated 15 inappropriate referrals corresponding to a total of 16 waiting days. The device reduced this to 11 inappropriate referrals with a cumulative waiting time of 18 days. The very small number of events in this subgroup prevents any conclusion on waiting-time direction; this subgroup analysis is exploratory.
Waiting list (secondary, supporting)
Within the analytical sample, the average waiting time for dermatologist consultation was 11.5 days, with observed waits ranging from 1 day to 61 days.
Under the assumption that the medical workforce remains constant, the observed 38% reduction in inappropriate referrals corresponds to an average waiting-time reduction from 11.5 days to approximately 7.1 days. Under the alternative assumption derived from the observed 56% reduction in cumulative unnecessary waiting time, the corresponding estimate is a reduction from 11.5 days to approximately 5.0 days. The conservative 7.1-day estimate is used as the primary figure; the 5.0-day estimate is reported as an upper bound. Both estimates are subject to the workforce-constant assumption and are not extrapolated beyond the analytical sample.
Within the analytical sample the device also exhibited higher sensitivity for referral-appropriateness than the primary care practitioner, which in the context of time-critical conditions (for example, aggressive melanoma) is clinically meaningful but requires confirmation in larger cohorts.
Illustrative individual cases are reported for completeness. Patient 23 was initially referred with a presumptive diagnosis of a nevus with irregular borders and was subsequently diagnosed with amelanotic melanoma at specialist review; the device flagged a moderate-to-high malignancy score on one of the clinical images, consistent with earlier referral. Patient 45 waited 37 days for a diagnosis of granuloma vs. a possible basal-cell carcinoma; the device assigned a high malignancy score consistent with basal-cell carcinoma. In total, three of 51 referred patients (approximately 6%) in the analytical sample waited longer than one month with concerns for potential skin cancer. These individual cases are reported as illustrative of the clinical relevance of the referral-appropriateness claim and are not used for inference.
Adverse Events and Adverse Reactions to the Product
As a non-interventional observational clinical investigation, participants did not interact directly with the device and did not undergo device-related physical procedures. The device was evaluated as a decision-support tool on images collected prospectively under the CIP and scored retrospectively under the manufacturer's configuration-management procedure.
Adverse-event surveillance was conducted by the Principal Investigators by passive reporting at each site, covering the period from the first subject's enrolment (2022-11-23) to the final data-lock date. No adverse events or adverse reactions related to the device were reported during the investigation. For the three patients who waited longer than one month with concerns for potential skin cancer (including one aggressive melanoma), review of the clinical outcome record found no reportable device-related harm; any delay in diagnosis for these patients is attributable to the primary-care referral pathway as it operated without the device and is considered under the benefit-risk assessment of the referral workflow rather than as a device-related adverse event. Product deficiencies are separately reported under §Product Deficiencies.
Product Deficiencies
No deficiencies in the product have been observed during the course of this study. As a result, no corrective actions have been deemed necessary. The product has demonstrated consistent performance in accordance with the study's objectives.
Subgroup Analysis for Special Populations
In the context of the analysed conditions, no special population subgroups were identified for this study. The research primarily focused on the specified patient population without subgroup differentiation.
Accounting for All Subjects
A total of 198 lesions and 127 patients were enrolled. Ten patients were excluded from the analysis due to lack of diagnostic confirmation. Consequently, the final analytical sample comprised 117 patients with a total of 184 images.
Discussion and Overall Conclusions
Clinical Performance, Efficacy and Safety
Summary of performance claims
studyCode or folderSlug prop, or ensure this component is used within an Investigation document with a registered folder slug.In the analytical sample, primary care practitioners exhibited a sensitivity of 45.2%: 14/31 (95% CI: [29.2%-62.2%]) for the decision to refer a patient to secondary care, and a specificity of 47.1%: 72/153 (95% CI: [39.3%-54.9%]). These figures are consistent with prior literature reporting limited sensitivity of primary-care referral decisions for dermatology.
Within the analytical sample, the most frequently referred conditions were common and relatively easy to diagnose when image-quality is adequate — actinic keratosis (18%), seborrheic keratosis (12%), and conditions such as psoriasis, erythema and eczema. The device correctly flagged these common conditions at the applied operating threshold, which within the sample supports the mechanism underlying the observed reduction in inappropriate referrals.
Image quality influences device performance, consistent with the DIQA-stratified results reported in §Impact of image quality. This is a known dependency for image-based decision support; the PMCF Plan identifies a pre-specified operating-quality threshold as a confirmatory activity.
Within the analytical sample, the primary endpoint was met: the device achieved a 38% reduction in inappropriate referrals, comfortably exceeding the pre-specified 15% MCID. The 95% confidence interval around the referral-rate difference excludes the null at α = 0.05 within the analytical sample. The analytical sample (117 patients / 184 images) is 49.5% of the pre-specified patient target and was achieved after the recruitment shortfall declared in §Protocol Deviations; the observed primary-endpoint effect size is sufficiently large in absolute terms that the observed 95% CI excludes the null within the analytical sample, but generalisation of the primary finding beyond the analytical sample is deferred to the independent-sample confirmation identified in the PMCF Plan. The analytical sample included a wide spectrum of dermatological pathologies — including melanocytic and non-melanocytic malignancies, pre-malignant keratoses and inflammatory dermatoses — and within the analytical sample the device's referral-appropriateness performance was consistent across subgroups at the level of precision the sample allows.
Benefit-risk assessment against GSPR 1 and GSPR 8
GSPR 1 (benefits outweigh risks). Within the analytical sample, the device reduced the number of inappropriate referrals from 81 to 50 (a 38% reduction) while achieving higher sensitivity for referral-appropriate cases than the primary care practitioner (74.2% vs. 45.2% at the applied operating threshold). At image level, the residual false-negative rate for referral-appropriateness was 8/31 (25.8%) within the analytical sample. Mitigations for the residual false-negative rate comprise: (i) the device's decision-support framing, under which the clinician retains final responsibility for the referral decision; (ii) the Top-5 prioritised differential output that enables the clinician to observe flagged differentials even where the binary referral decision is negative; and (iii) the safety-information content of the IFU.
GSPR 8 (use-error risk). No adverse events or device-related harms were observed during the investigation. The principal use-error risk identified by the risk-management record is misinterpretation of the device's output as a diagnostic determination rather than decision support. Mitigations comprise: (i) the IFU decision-support framing; (ii) the IFU integration-requirements section, which mandates that the integrating system display the Top-5 prioritised differential, the malignancy gauge and the referral recommendation to the clinician; and (iii) the device's continuous monitoring of image quality via the DIQA algorithm, which prevents scoring of images below the run-time quality gate.
Residual risks for both GSPR 1 and GSPR 8 are tracked in R-TF-013-002 Risk Management Record and are reviewed under the manufacturer's post-market surveillance procedure.
Clinical Risks and Benefits
Participants in this investigation did not undergo any procedures that posed a risk to their safety. Using the device supports the primary care practitioner in optimising referral decisions, which within the analytical sample reduced inappropriate referrals and cumulative unnecessary waiting time.
Clinical Relevance
The device supports the primary care practitioner's referral decision by providing image-based decision support grounded in machine-vision and deep-learning techniques validated in the peer-reviewed literature9 10 11 12. This approach is aligned with the body of research on the integration of artificial intelligence and machine learning in dermatological decision support13 14.
Prior studies have demonstrated the potential of machine-learning algorithms in diagnosing dermatological conditions including acne, nevi, basal cell carcinoma and psoriasis15 16. The device's applicability to remote consultation addresses a recognised need in modern healthcare17.
Within the analytical sample, the device's referral-appropriateness performance against the primary care practitioner supports the use of the device as decision support in the primary-care referral pathway; the observed reduction in inappropriate referrals and the observed reduction in cumulative unnecessary waiting time are consistent with published evidence that validated triage tools reduce inappropriate specialist consultations and improve access for patients who genuinely require specialist care18 19 20.
Malignancy detection is reported as a secondary, exploratory analysis in this investigation. The observed AUC of 0.815 and NPV of 96.4% within the analytical sample indicate discriminatory ability in the sample, but the 95% CI lower bound for malignancy sensitivity (32.6%) is insufficient to support a screening or rule-out claim on the basis of this investigation alone. A rule-out or screening claim requires confirmation in a dedicated, powered study; this is committed to under the PMCF Plan. Published evidence independently supports the clinical value of AI-based decision support for malignancy detection21 22 and the impact of earlier detection on skin-cancer outcomes23 24.
The reduction in consultation time and the patient-centred framing of the device are consistent with broader trends in efficient, patient-centric healthcare delivery25 26. The absence of device-related adverse events observed in this investigation is consistent with current standards for medical-device safety27.
The device combines decision support with continuous image-quality monitoring (DIQA), distinguishing it from tools focused solely on diagnostic classification28 29.
In summary, within the analytical sample the device supports the primary care practitioner's referral decision, reduces inappropriate referrals and reduces cumulative unnecessary waiting time. Independent-sample confirmation of the primary endpoint at a pre-specified operating threshold, and confirmation of malignancy-detection performance in a dedicated powered study, are identified in the PMCF Plan.
Specific Benefit or Special Precaution
Benefits
- The device supports diagnostic decision-making across a wide range of skin lesions from digital images.
- Automated decision support provides rapid feedback to the healthcare practitioner, easing and accelerating clinical practice.
- The device supports optimisation of referrals and teledermatology, reducing waiting lists and associated costs and improving the treatment and experience of the patient.
- The device also evaluates the severity of several dermatological conditions, which supports monitoring of disease progression and treatment effectiveness and reduces the time burden on the medical practitioner.
Precautions
- The device must be used as a clinical support and not to replace the expertise of the medical practitioner.
- The device can only analyse visible lesions and provide insight into a closed set of skin lesions. Skin lesions not learnt by the device can not be diagnosed.
- Images taken at low quality can lead to unreliable output. To manage this dependency, the device incorporates the DIQA11 algorithm, which continuously monitors image quality at run time and prevents scoring of images below the run-time quality gate.
Implications for Future Research
The observations from this investigation identify several follow-on activities under the PMCF Plan (R-TF-007-002):
- Independent-sample confirmation of the primary endpoint at a pre-specified operating threshold, in a powered cohort representative of the intended European deployment settings.
- Dedicated, powered confirmation of malignancy-detection performance, including melanoma and non-melanocytic skin cancer, in a cohort sufficient to establish the lower bound of sensitivity at a pre-specified operating threshold.
- A pre-specified operating-quality (DIQA) threshold for the device's referral output, selected independently of the present sample.
- Phototype-coverage generalisation to Fitzpatrick V and VI populations (addressed by dedicated phototype-bridging evidence at Clinical Evaluation level and by the PMCF Plan).
- Longer-term outcome and health-economic activities to quantify patient-outcome and cost-impact under documented assumptions.
Limitations of Clinical Research
The performance of image-based decision support depends on image-quality; variability in illumination, colour, shape, size and focus, and a limited number of images per patient, can reduce diagnostic accuracy. This dependency is partially managed by the DIQA run-time quality gate.
The recruitment shortfall declared in §Protocol Deviations reduces the precision of secondary and subgroup estimates, particularly for less-prevalent conditions and for malignancy subgroups. The primary endpoint was met within the analytical sample, but confirmation in an independent, pre-specified-threshold sample is required to support generalisation beyond the analytical sample.
The referral-decision threshold of 0.45 applied in the primary analysis was informed by a Youden-J sweep on the analytical sample and is therefore subject to optimistic bias; this is declared in §Protocol Deviations and is to be addressed by the PMCF confirmatory activity.
Malignancy detection is reported as secondary and exploratory; the number of malignant cases (n = 14) is insufficient to support a confirmatory claim and the lower bound of the 95% CI for malignancy sensitivity (32.6%) does not support a screening or rule-out claim on the basis of this investigation alone.
All participants were recruited from primary care in the Basque Country (Spain); generalisability to other European healthcare systems, patient populations and primary-care workflows remains to be established via the PMCF activities. The analytical cohort is predominantly Fitzpatrick I–III and does not, on its own, support performance claims on Fitzpatrick V or VI populations.
Ethical Aspects of Clinical Research
This study adhered to international Good Clinical Practice (GCP) guidelines, the Declaration of Helsinki in its latest amendment, and applicable international and national regulations. As applicable, approval from the relevant Ethics Committee was obtained prior to the initiation of the study. When applicable, modifications to the protocol were reviewed and approved by the Principal Investigator (PI) and subsequently evaluated by the Ethics Committee before subjects were enrolled under a modified protocol.
This study was conducted in compliance with European Regulation 2016/679, of 27 April, concerning the protection of natural persons with regard to the processing of personal data and the free movement of such data (General Data Protection Regulation, GDPR), and Organic Law 3/2018, of 5 December, on the Protection of Personal Data and the guarantee of digital rights. In accordance with these regulations, no data enabling the personal identification of participants was collected, and all information was managed securely in an encrypted format.
Participants were informed both orally and in writing about all relevant aspects of the study, with the information being tailored to their level of understanding. They were provided with a copy of the informed consent form and the accompanying patient information sheet. Adequate time was given to patients to ask questions and fully comprehend the details of the study before providing their consent.
The PI was responsible for the preparation of the informed consent form, ensuring it included all elements required by the International Conference on Harmonisation (ICH), adhered to current regulatory guidelines, and complied with the ethical principles of GCP and the Declaration of Helsinki.
The original signed informed consent forms were securely stored in a restricted access area under the custody of the PI. These documents remained at the research site at all times. Participants were provided with a copy of their signed consent form for their records.
Investigators and Administrative Structure of Clinical Research
Brief Description
The clinical-investigation team comprises experienced dermatologists and the manufacturer's technical-support staff. Dr. Jesús Gardeazabal García and Dr. Rosa María Izu Belloso served as Principal Investigators, affiliated with Osakidetza – Servicio Vasco de Salud. Technical support for the investigation was provided by the manufacturer's Chief Technology Officer and Chief Executive Officer. This team ensured a comprehensive approach to the clinical evaluation of the device in a real-world primary-care setting comprising the Sodupe-Güeñes, Balmaseda, Buruaga and Zurbaran health centres.
Investigators
Principal investigators
- Dr. Jesús Gardeazabal García (Hospital Universitario Cruces)
- Dr. Rosa Mª Izu Belloso (Hospital Universitario Basurto)
Technical Support (Manufacturer)
- Mr. Alfonso Medela — Chief Technology Officer
- Mr. Taig Mac Carthy — Chief Executive Officer
Centres
- Health Centre Sodupe-Güeñes
- Health Centre Balmaseda
- Health Centre Buruaga
- Health Centre Zurbaran
External Organisation
No additional organisations, beyond those previously mentioned, contributed to the clinical research. The investigation was conducted with the collaboration and resources of the specified entities.
Sponsor and Monitor
The sponsor and monitor of the investigation is the manufacturer identified in §Sponsor Identification and Contact.
Report Annexes
- The Ethics Committee resolution (CEIm of Euskadi, reference PS2022074, 2022-11-23, together with its substantial modification) is retained in the Trial Master File and available to the notified body and to competent authorities on formal request.
- The Instructions for Use (IFU) applicable to the investigation is provided alongside the Clinical Investigation Plan.
Signature meaning
The signatures for the approval process of this document can be found in the verified commits at the repository for the QMS. As a reference, the team members who are expected to participate in this document and their roles in the approval process, as defined in Annex I Responsibility Matrix of the GP-001, are:
- Author: Team members involved
- Reviewer: JD-018 Clinical Research Coordinator
- Approver: JD-022 Medical Manager