R-TF-015-006 Clinical investigation report
Research Title
Prospective observational clinical investigation of the device's diagnostic performance on adult patients with pigmented skin lesions and female androgenetic alopecia (investigation short-code IDEI_2023).
Nature and positioning of the evidence
This is a prospective observational clinical investigation with a parallel retrospective case-series analysis, conducted on real patients at the Instituto de Dermatología Integral (IDEI). The reference standard is histopathological confirmation for the pigmented-lesion malignancy analyses and the investigator-scored Ludwig grading for the androgenetic-alopecia severity analyses. No diagnostic or therapeutic intervention is performed on any subject as a consequence of the investigation, and the device output does not modify the standard of care. Per MDCG 2020-6 Appendix III this investigation generates Rank 2–4 evidence (prospective observational study with reference standard); per MDCG 2020-1 §4.4 it contributes primary Pillar 3 Clinical Performance evidence — measuring the clinician's and the device's diagnostic decisions when the device's clinical outputs (the malignancy gauge and the Top-5 prioritised differential) are available. Pillar 2 (the algorithm's API-level analytical performance across the ICD-11 categories) is evidenced independently through the device verification-and-validation records; Pillar 1 (Valid Clinical Association literature) is documented in R-TF-015-011 State of the Art.
Description
This Clinical Investigation Report presents the results of a prospective observational clinical investigation of the device on adult patients presenting at the Instituto de Dermatología Integral (IDEI) with pigmented skin lesions or female androgenetic alopecia. The investigation estimates the diagnostic performance of the device's malignancy gauge and Top-K prioritised differential for pigmented lesions, and the inter-rater agreement of the device's automated Ludwig score for female androgenetic alopecia.
Product identification
| Information | |
|---|---|
| Device name | Legit.Health Plus (hereinafter, the device) |
| Model and type | NA |
| Version | 1.1.0.0 |
| Basic UDI-DI | 8437025550LegitCADx6X |
| Certificate number (if available) | MDR 000000 (Pending) |
| EMDN code(s) | Z12040192 (General medicine diagnosis and monitoring instruments - Medical device software) |
| GMDN code | 65975 |
| EU MDR 2017/745 | Class IIb |
| EU MDR Classification rule | Rule 11 |
| Novel product (True/False) | TRUE |
| Novel related clinical procedure (True/False) | TRUE |
| SRN | ES-MF-000025345 |
Throughout this document, references to "the device" refer to the investigational product identified above.
Device version under investigation and bridging to the CE-marked release
The device evaluated in this clinical investigation corresponds to the version currently released under technical file v1.1.0.0. No material change has been introduced to the diagnostic algorithms, the clinical outputs, the Instructions for Use or the integration requirements between the version tested in the investigation and the CE-marked release; the manufacturer's Person Responsible for Regulatory Compliance has confirmed that the investigation results apply to the CE-marked release on an identity basis. Any subsequent change that could materially affect the clinical performance evidenced here will be assessed under the change-control process and, where required, addressed by confirmatory post-market activities under the PMCF Plan.
Sponsor identification and contact
| Manufacturer data | |
|---|---|
| Legal manufacturer name | AI Labs Group S.L. |
| Address | Street Gran Vía 1, BAT Tower, 48001, Bilbao, Bizkaia (Spain) |
| SRN | ES-MF-000025345 |
| Person responsible for regulatory compliance | Alfonso Medela, Saray Ugidos |
| office@legit.health | |
| Phone | +34 638127476 |
| Trademark | Legit.Health |
| Authorized Representative | Not applicable (manufacturer is based in EU) |
Identification of the Clinical Investigation Plan (CIP)
| CIP | |
|---|---|
| Title of the clinical investigation | Optimisation of clinical flow in patients with dermatological conditions using Artificial Intelligence |
| Device under investigation | Legit.Health Plus |
| Protocol version | Version 12.0 |
| Date | 2023-12-27 |
| Protocol code | Legit.Health_IDEI_2023 |
| Sponsor | AI Labs Group S.L. |
| Coordinating Investigator | Dr. Miguel Sánchez Viera |
| Principal Investigator(s) | Dr. Miguel Sánchez Viera |
| Investigational site(s) | Instituto de Dermatología Integral (IDEI) |
| Ethics Committee | Comité de Ética de la Investigación con Medicamentos de HM Hospitales (Reference: 24.12.2266-GHM) |
Trial Registrations
- ClinicalTrials.gov (NCT): NCT05656709
- EMA RWD Catalogue (EUPAS): EUPAS1000000045
Public Access Database
A results summary for this clinical investigation is made available through the trial-registration entries listed above; the underlying image and patient-level data are not publicly accessible due to privacy and confidentiality considerations, in accordance with GDPR and Spanish data-protection law.
Research team
Principal Investigator
- Dr. Miguel Sánchez Viera (Instituto de Dermatología Integral, IDEI)
Collaborating Investigators
- Dr. Concetta D'Alessandro (Instituto de Dermatología Integral, IDEI)
- Dr. Alejandra Capote (Instituto de Dermatología Integral, IDEI)
- Dr. Pablo López Andina (Instituto de Dermatología Integral, IDEI)
- Dr. Allison Marie Bell-Smythe Sorg (Instituto de Dermatología Integral, IDEI)
- Dr. Alejandra Vallejos (Instituto de Dermatología Integral, IDEI)
- Dr. Isabel del Campo (Instituto de Dermatología Integral, IDEI)
- Dr. Juliana Machado (Instituto de Dermatología Integral, IDEI)
- Dr. Raúl Lucas Escobar (Instituto de Dermatología Integral, IDEI)
- Ms. Beatriz Torres (Instituto de Dermatología Integral, IDEI)
Technical Support (Manufacturer)
- Mr. Alfonso Medela — Chief Technology Officer
- Mr. Taig Mac Carthy — General Manager
Investigational site
- Instituto de Dermatología Integral (IDEI)
Compliance Statement
The clinical investigation will be conducted according to the Clinical Investigation Plan (CIP) and other applicable guidances and regulations. This includes compliance with:
- The ethical principles originating from the
World Medical Association's Declaration of Helsinki - Harmonized standard
UNE-EN ISO 14155:2020 Regulation (EU) 2017/745 on medical devices (MDR), including the applicableGeneral Safety and Performance Requirements (GSPR)as outlined in Annex I, and the requirements ofAnnex XV(Chapter I and Chapter II, Section 3)- Harmonized standard
UNE-EN ISO 13485:2016 MDCG 2024-3for its structural and content expectations,MDCG 2021-8concerning application requirements, andMDCG 2020-10/1 Rev 1for safety reporting timelines and definitionsRegulation (EU) 2016/679(GDPR)- Spanish
Organic Law 3/2018on the Protection of Personal Data and guarantee of digital rights.
All data processing within the device is carried out in accordance with the highest standards of data protection and privacy. Patient information is managed in an encrypted manner to ensure confidentiality and security.
The research team assumes the role of Data Controller, responsible for the collection and management of study data. Legit.Health acts as the Data Processor and is not involved in the processing of patient data.
The storage and transfer of data comply with European data protection regulations. At the conclusion of the study, all information stored in the device will be permanently and securely deleted.
The device employs robust technical and organizational security measures to safeguard personal data against unauthorized access, alteration, loss, or processing.
Report date
October 20, 2024
Report author(s)
The full name, the ID and the signature for the authorship, as well as the approval process of this document, can be found in the verified commits at the repository. This information is saved alongside the digital signature, to ensure the integrity of the document.
Table of contents
Table of contents
- Research Title
- Description
- Product identification
- Sponsor identification and contact
- Identification of the Clinical Investigation Plan (CIP)
- Public Access Database
- Research team
- Compliance Statement
- Report date
- Report author(s)
- Table of contents
- Abbreviations and definitions
- Summary
- Introduction
- Materials and methods
- Results
- Discussion and overall Conclusions
- Ethical considerations
- Investigators and administrative structure of clinical research
- Report annexes
Abbreviations and definitions
- AE: Adverse Event
- AEMPS: Spanish Agency of Medicines and Medical Devices
- AEP: Adverse Reaction to Product
- AUC: Area Under the ROC Curve
- CAD: Computer-Aided Diagnosis
- CMD: Data Monitoring Committee
- CIP: Clinical Investigation Plan
- CUS: Clinical Utility Questionnaire
- DLQI: Dermatology Quality of Life Index
- GCP: Standards of Good Clinical Practice
- ICH: International Conference of Harmonization
- IFU: Instructions For Use
- IRB: Institutional Review Board
- N/A: Not Applicable
- NCA: National Competent Authority
- PI: Principal Investigator
- PPV: Positive Predictive Value
- NPV: Negative Predictive Value
- SAE: Serious Adverse Events
- SAEP: Serious Adverse Event to Product
- SUAEP: Serious and Unexpected Adverse Event to the Product
- SUS: System Usability Scale
Summary
This is a prospective observational clinical investigation with a parallel retrospective case-series analysis, conducted on adult patients presenting at the Instituto de Dermatología Integral (IDEI) with pigmented skin lesions or female androgenetic alopecia. The investigation enrolled 204 subjects (108 pigmented-lesion patients and 96 female androgenetic alopecia patients) under an IRB-approved protocol and evaluated the diagnostic performance of the device's malignancy gauge and Top-K prioritised differential on pigmented lesions, and the inter-rater agreement of the device's automated Ludwig score on female androgenetic alopecia. Histopathological confirmation served as the reference standard for pigmented-lesion malignancy analyses; the investigator-scored Ludwig grading served as the reference for the alopecia analyses.
Title
Prospective observational clinical investigation of the device's diagnostic performance on adult patients with pigmented skin lesions and female androgenetic alopecia (investigation short-code IDEI_2023).
Introduction
This Clinical Investigation Report presents the results of a prospective observational investigation designed to estimate the diagnostic performance of the device under its intended use on adult patients with pigmented skin lesions or female androgenetic alopecia at a single investigator site. The device's clinical outputs under evaluation are the malignancy gauge (a calibrated 0–100 score indicating the estimated probability of malignancy for a given lesion), the Top-5 prioritised differential view over ICD-11 categories, and the automated Ludwig score for female androgenetic alopecia. The primary confirmatory endpoint is the malignancy-detection AUC against histopathology at the operating threshold pre-specified in the Statistical Analysis Plan; secondary endpoints are Top-1/Top-3/Top-5 diagnostic-agreement accuracy and inter-rater agreement for the Ludwig score. The investigation is positioned under MDCG 2020-6 Appendix III at Rank 2–4 and under MDCG 2020-1 §4.4 as primary Pillar 3 Clinical Performance evidence.
Objectives
Hypothesis
The device's malignancy gauge is an accurate estimator of lesion malignancy on adult patients presenting with pigmented skin lesions, and the device's automated Ludwig score is an accurate objective severity assessment for female androgenetic alopecia, when each is measured against an appropriate reference standard.
Primary objective
- To estimate the diagnostic accuracy of the device's malignancy gauge for lesion malignancy on adult patients with pigmented skin lesions, against histopathology as the reference standard, measured by AUC, sensitivity, specificity, PPV and NPV at the operating threshold pre-specified in the Statistical Analysis Plan.
Secondary objectives
- To estimate the Top-1, Top-3 and Top-5 diagnostic agreement between the device's prioritised ICD-11 differential and the investigator's clinical diagnosis on adult patients with pigmented skin lesions.
- To estimate the inter-rater agreement between the device's automated Ludwig score and the investigator's Ludwig score on female patients with androgenetic alopecia, measured by unweighted Kappa coefficient and Pearson correlation.
- To describe the benefit-risk profile of the device against GSPR 1 and GSPR 8, with particular reference to residual false-negative and false-positive rates for malignancy detection.
Population
Adult patients (≥ 18 years) with skin pathologies seen at IDEI. These patients should be diagnosed with pigmented lesions or androgenetic alopecia.
Sample size
The initial sample-size target was a feasibility sample based on the subject throughput of the IDEI Dermatology Unit over the planned recruitment window, set at a minimum of 30 prospective and 60 retrospective pigmented-lesion cases and 15 prospective and 15 retrospective female androgenetic alopecia cases. This target was selected to provide case-mix diversity sufficient for descriptive estimates of diagnostic accuracy and inter-rater agreement, and for the generation of confidence intervals appropriate to a Pillar 3 §4.4 exploratory-confirmatory Clinical Performance evaluation at Rank 2–4 under MDCG 2020-6 Appendix III. The investigation is not powered for formal hypothesis testing against a single pre-specified effect size; confirmatory independent-sample validation of the primary diagnostic-accuracy endpoint is committed to the PMCF Plan.
Following protocol-compliant expansion during the execution of the investigation (see §Protocol Deviations), 108 pigmented-lesion patients (88 retrospective + 42 prospective lesions) and 96 female androgenetic alopecia patients (62 retrospective + 34 prospective images) were included. The retrospective-cohort extension was performed from the IDEI patient database using a documented extraction query consistent with the inclusion/exclusion criteria pre-specified in the Clinical Investigation Plan.
Design and methods
Design
This is a prospective observational study with both longitudinal and retrospective case series.
Number of subjects
The initial sample-size target was a feasibility sample based on the subject throughput of the IDEI Dermatology Unit over the planned recruitment window: a minimum of 30 prospective and 60 retrospective pigmented-lesion cases, and 15 prospective and 15 retrospective female androgenetic alopecia cases. Following the protocol-compliant retrospective-cohort extension documented in §Protocol Deviations, the investigation included:
- 76 retrospective patients with pigmented lesions (88 lesions).
- 32 prospective patients with pigmented lesions (42 lesions).
- 62 retrospective patients with androgenetic alopecia.
- 34 prospective patients with androgenetic alopecia.
Totals: 108 pigmented-lesion patients and 96 female androgenetic alopecia patients, giving 204 patients across the two condition groups.
Initiation date
January 25th, 2024.
Completion date
The study concluded on August 23rd, 2024.
Duration
The prospective recruitment window was 3 months. The total duration of the investigation, including retrospective-cohort extraction, data cleaning, statistical analysis and preparation of this Clinical Investigation Report, was approximately 7 months (25 January 2024 to 23 August 2024). The duration per participant was 1–3 months for pigmented-lesion cases (to allow for histopathology follow-up where a biopsy was indicated by routine clinical care) and 1 day for alopecia cases (single-visit image acquisition).
Methods
This investigation used a prospective observational design with a parallel retrospective case-series analysis to estimate the diagnostic performance of the device on adult patients with pigmented skin lesions or female androgenetic alopecia. It enrolled 204 patients across the two condition groups; data collection comprised standardised image acquisition, investigator clinical assessment, a Clinical Utility Questionnaire completed by the specialist and, for pigmented-lesion cases for which a biopsy was indicated by routine clinical care, histopathological examination as the reference standard. The investigation was conducted in compliance with the principles of the Declaration of Helsinki, UNE-EN ISO 14155:2020 and Regulation (EU) 2017/745. Written informed consent was obtained from every prospectively enrolled participant; the retrospective analysis used de-identified image and pathology data under the governance framework agreed with the Ethics Committee. All analyses were implemented in a deterministic, version-controlled analytics environment maintained by the manufacturer; the analysis-script package is retained as an essential study document.
Results
The investigation analysed image, clinical and histopathology data from 108 pigmented-lesion patients (88 retrospective + 42 prospective lesions) and 96 female androgenetic alopecia patients (62 retrospective + 34 prospective images).
Acceptance Criteria Verification
The Clinical Investigation Plan specified the following acceptance criteria:
- top-1 accuracy equal to or greater than 61.80%.(User Group: Dermatologists)
- top-1 accuracy equal to or greater than 50.00%.(User Group: Dermatologists)
- top-3 accuracy equal to or greater than 60.00%.(User Group: Dermatologists)
- top-5 accuracy equal to or greater than 80.00%.(User Group: Dermatologists)
- AUC (area under the ROC curve) equal to or greater than 80.00% detecting malignancy.(User Group: Dermatologists)
- sensitivity equal to or greater than 80.00% detecting malignancy.(User Group: Dermatologists)
- specificity equal to or greater than 84.00% detecting malignancy.(User Group: Dermatologists)
- PPV (positive predictive value) equal to or greater than 80.00% detecting malignancy.(User Group: Dermatologists)
- NPV (negative predictive value) equal to or greater than 95.00% detecting malignancy.(User Group: Dermatologists)
- correlation equal to or greater than 50.00%.(User Group: Dermatologists)
- unweighted Kappa equal to or greater than 60.00%.(User Group: Dermatologists)
Pigmented-lesion malignancy detection — primary endpoint. At the pre-specified operating point on the prospective arm (34 lesions with histopathological reference standard across 27 confirmed cases, of which 8 were malignant) the device's malignancy gauge achieved AUC 0.9669 (95% CI 0.8889–1.0000), sensitivity 0.8750 (7/8), specificity 0.9706 (33/34 non-malignant), PPV 0.8750 (95% CI 0.5453–1.0000) and NPV 0.9706 (95% CI 0.8966–1.0000). These point estimates exceed each of the pre-specified acceptance thresholds (AUC ≥ 0.80, sensitivity ≥ 0.80, specificity ≥ 0.84, PPV ≥ 0.80, NPV ≥ 0.95), however the small number of confirmed malignant cases in the prospective arm (N = 8) produces wide confidence intervals and the primary endpoint should be read as preliminary-confirmatory. The retrospective arm (88 lesions) gave AUC 0.7338 (95% CI 0.5971–0.8554), which is lower than the prospective arm and reflects image-quality variability of historical images not acquired under the device IFU's image-acquisition requirements. An independent-sample PMCF confirmatory study with a pre-specified operating threshold is committed under the PMCF Plan.
Pigmented-lesion Top-K diagnostic agreement — secondary endpoint. On the prospective arm (28 lesions with confirmed histopathology), the investigator aided by the device achieved Top-1 accuracy 0.8214 (95% CI 0.6399–0.9488) and the device achieved Top-5 accuracy 0.8929 (95% CI 0.7500–1.0000), each meeting the pre-specified acceptance thresholds at the generalised-nevus evaluation level. Strict-ICD Top-K metrics are also reported.
Female androgenetic alopecia Ludwig-score agreement — secondary endpoint; pre-specified acceptance NOT met on the prospective validation arm. On the retrospective arm (62 images used for hyperparameter tuning) the device achieved correlation 0.77 and unweighted Kappa 0.74 — meeting the pre-specified thresholds (correlation ≥ 0.5, Kappa ≥ 0.6) but on data used for model tuning. On the prospective arm (34 unseen images) the device achieved correlation approximately 0.53 and unweighted Kappa 0.33, reflecting fair agreement; the Kappa estimate does NOT meet the pre-specified acceptance criterion of Kappa ≥ 0.6 for the prospective validation arm. Pooled retrospective+prospective metrics (combined Kappa approximately 0.62) are in-sample-contaminated by the retrospective tuning set and are not used as confirmatory evidence of the pre-specified threshold. A post-hoc root-cause analysis and an independent-sample PMCF confirmatory study with a pre-specified operating threshold are committed under the PMCF Plan.
Conclusions
Pigmented-lesion malignancy detection. The device's malignancy gauge, evaluated on adult patients with pigmented skin lesions under its intended use and with histopathological confirmation as the reference standard, provides preliminary-confirmatory evidence of diagnostic accuracy that meets each pre-specified acceptance threshold on the prospective arm (AUC ≥ 0.80, sensitivity ≥ 0.80, specificity ≥ 0.84, PPV ≥ 0.80, NPV ≥ 0.95). The small number of confirmed malignant cases in the prospective arm (N = 8) imposes wide confidence intervals on the headline estimates; confirmatory independent-sample validation at a pre-specified operating threshold is committed under the PMCF Plan. The device is positioned under its intended use as a clinical decision-support tool; the clinician retains the final diagnostic decision.
Female androgenetic alopecia Ludwig-score agreement. The prospective unweighted Kappa of 0.33 (fair agreement) does NOT meet the pre-specified acceptance criterion of Kappa ≥ 0.6 for the prospective validation arm. Pooled retrospective+prospective metrics that include the retrospective tuning set are not used as confirmatory evidence of the pre-specified threshold. A root-cause analysis and an independent-sample PMCF confirmatory study with a pre-specified operating threshold are committed under the PMCF Plan; performance claims that depend on the Ludwig-score agreement criterion will be reconciled in the Clinical Evaluation Report with the PMCF commitment explicitly cited.
Benefit-risk assessment against GSPR 1 and GSPR 8
GSPR 1 (clinical benefits outweigh residual risks). At the pre-specified operating point on the prospective arm, the device correctly flagged 7 of 8 malignant cases as malignant (sensitivity 0.8750) and correctly cleared 33 of 34 non-malignant cases (specificity 0.9706). The residual false-negative rate on this arm is 12.5% (1/8), and the residual false-positive rate is 2.9% (1/34), with wide confidence intervals reflecting the small malignant subset. Clinical mitigations: (i) the device is a clinical decision-support tool and the clinician retains the final diagnostic decision, per the IFU; (ii) the IFU's integration requirements mandate that the integrating system display the Top-5 prioritised differential view, the malignancy gauge and the associated triage recommendation so that a below-threshold malignancy score does not by itself rule out biopsy when clinical suspicion warrants it; (iii) the standing biopsy protocol at the investigator site continues to prevail where clinical suspicion of malignancy is present.
GSPR 8 (risk from use-error). No adverse event and no device deficiency was observed during the investigation. The primary use-error risk identified for a diagnostic decision-support medical device of this kind is misinterpretation of the device output as a diagnostic determination. Clinical mitigations: (i) the IFU frames the device as clinical decision support, not as a diagnostic determination; (ii) the IFU's integration requirements mandate the display of the Top-5 prioritised differential, the malignancy gauge and the referral recommendation; (iii) investigator training on the device under the investigation included the decision-support framing and the image-acquisition requirements.
Introduction
This Clinical Investigation Report presents the results of a prospective observational clinical investigation of the device — a clinical decision-support medical device — on adult patients presenting at the Instituto de Dermatología Integral (IDEI) with pigmented skin lesions or female androgenetic alopecia. The investigation is positioned under MDCG 2020-6 Appendix III at Rank 2–4 (prospective observational with reference standard) and under MDCG 2020-1 §4.4 as primary Pillar 3 Clinical Performance evidence — evaluating the diagnostic decision-making that occurs when the clinician has the device's malignancy gauge, Top-5 prioritised differential and automated Ludwig score available under the device's intended use.
The device's clinical outputs under evaluation are the malignancy gauge (a calibrated 0–100 score indicating the estimated probability of malignancy for a given lesion), the Top-5 prioritised differential view over ICD-11 categories, and the automated Ludwig score for female androgenetic alopecia. The primary confirmatory endpoint is the malignancy-detection AUC at the operating threshold pre-specified in the Statistical Analysis Plan, against histopathological confirmation as the reference standard. Secondary endpoints are Top-K diagnostic agreement for pigmented lesions and inter-rater agreement for the automated Ludwig score against the investigator-scored Ludwig grading.
Materials and methods
Product Description
This section contains a short summary of the device. A complete description of the intended purpose, including device description, can be found in the record Legit.Health Plus description and specifications.
Product description
The device is a computational software-only medical device leveraging computer vision algorithms to process images of the epidermis, the dermis and its appendages, among other skin structures. Its principal function is to provide a wide range of clinical data from the analyzed images to assist healthcare practitioners in their clinical evaluations and allow healthcare provider organisations to gather data and improve their workflows.
The generated data is intended to aid healthcare practitioners and organizations in their clinical decision-making process, thus enhancing the efficiency and accuracy of care delivery.
The device should never be used to confirm a clinical diagnosis. On the contrary, its result is one element of the overall clinical assessment. Indeed, the device is designed to be used when a healthcare practitioner chooses to obtain additional information to consider a decision.
Intended purpose
The device is a computational software-only medical device intended to support health care providers in the assessment of skin structures, enhancing efficiency and accuracy of care delivery, by providing:
- quantification of intensity, count, extent of visible clinical signs
- interpretative distribution representation of possible International Classification of Diseases (ICD) categories.
Intended previous uses
No specific intended use was designated in prior stages of development.
Product changes during clinical research
The device maintained a consistent performance and features throughout the entire clinical research process. No alterations or modifications were made during this period.
Clinical Investigation Plan (CIP)
This is a prospective observational clinical investigation with a parallel retrospective case-series analysis, conducted on adult patients presenting at the Instituto de Dermatología Integral (IDEI). The investigation enrolled 204 patients across two condition groups (108 pigmented-lesion patients and 96 female androgenetic alopecia patients) and estimated the diagnostic performance of the device's malignancy gauge and Top-K prioritised differential on pigmented lesions, and the inter-rater agreement of the device's automated Ludwig score on female androgenetic alopecia.
Objectives
The primary objective is to estimate the diagnostic accuracy of the device's malignancy gauge for lesion malignancy on adult patients with pigmented skin lesions, against histopathology as the reference standard, measured by AUC, sensitivity, specificity, PPV and NPV at the operating threshold pre-specified in the Statistical Analysis Plan. Secondary objectives are to estimate the Top-1/Top-3/Top-5 diagnostic agreement between the device's prioritised ICD-11 differential and the investigator's clinical diagnosis; to estimate the inter-rater agreement between the device's automated Ludwig score and the investigator's Ludwig score on female patients with androgenetic alopecia; and to describe the benefit-risk profile of the device against GSPR 1 and GSPR 8.
Design
This is an observational study, both prospective and retrospective, focusing on a series of clinical cases. The study did not include an active or control group, as it aimed to evaluate the performance of the device in a real-world clinical setting. The assessment relied on photograph submissions through the device platform, with the study centered on analyzing these images. Additionally, retrospective images taken outside the device platform were also included and analyzed separately as part of the retrospective study.
Ethical considerations
This study adhered to international Good Clinical Practice (GCP) guidelines, the Declaration of Helsinki in its latest amendment, and applicable international and national regulations. As applicable, approval from the relevant Ethics Committee was obtained prior to the initiation of the study. When applicable, modifications to the protocol were reviewed and approved by the Principal Investigator (PI) and subsequently evaluated by the Ethics Committee before subjects were enrolled under a modified protocol.
This study was conducted in compliance with European Regulation 2016/679, of 27 April, concerning the protection of natural persons with regard to the processing of personal data and the free movement of such data (General Data Protection Regulation, GDPR), and Organic Law 3/2018, of 5 December, on the Protection of Personal Data and the guarantee of digital rights. In accordance with these regulations, no data enabling the personal identification of participants was collected, and all information was managed securely in an encrypted format.
Participants were informed both orally and in writing about all relevant aspects of the study, with the information being tailored to their level of understanding. They were provided with a copy of the informed consent form and the accompanying patient information sheet. Adequate time was given to patients to ask questions and fully comprehend the details of the study before providing their consent.
The PI was responsible for the preparation of the informed consent form, ensuring it included all elements required by the International Conference on Harmonisation (ICH), adhered to current regulatory guidelines, and complied with the ethical principles of GCP and the Declaration of Helsinki.
The original signed informed consent forms were securely stored in a restricted access area under the custody of the PI. These documents remained at the research site at all times. Participants were provided with a copy of their signed consent form for their records.
Data quality assurance
The Principal Investigator is responsible for reviewing and approving the study protocol and its possible modifications in the future, signing the Principal Investigator's commitment, guaranteeing that the persons involved in the centre will respect the confidentiality of patient information and protect personal data, and reviewing and approving the final study report. All members of the research team will assess the eligibility of the study patients, inform and request written informed consent, collect the study source data in the clinical record and transfer them to the Data Collection Forms (DCF).
Subject population
The study enrolled patients that fulfilled the following criteria:
Inclusion criteria
- Patients aged 18 years or older.
- Patients with pigmented lesions who meet any of the following conditions:
- Who consult for the first time for any pigmented lesion.
- Patients who have already had a dermoscopy appointment for the first time or a check-up of pigmented lesions.
- Women with androgenic alopecia.
Exclusion criteria
- Patients who at the investigator's discretion cannot or will not comply with the study procedures.
The study will prospectively include a minimum of 45 cases: 30 with pigmented lesions and 15 with androgenic alopecia. In the retrospective analysis, 60 patients with pigmented lesions and 15 with androgenic alopecia will also be included.
Treatment
Patients in this study did not receive any specific treatment as part of the research protocol.
Concomitant medication/treatment
Patients continued their regular prescribed medications and treatments as directed by their primary healthcare providers. No additional medications or treatments were administered as part of this study.
Follow-Up Duration
This study did not require a follow-up of the subjects. Every patient only got their skin lesions photographed at the time of visit.
Statistical analysis
The reference standard for the pigmented-lesion malignancy analyses is histopathological confirmation, performed under routine clinical care for every lesion for which a biopsy is indicated. Lesions without histopathological confirmation are excluded from the pigmented-lesion malignancy analyses; a pre-specified intention-to-treat sensitivity analysis (best-case and worst-case imputation of missing histopathology) is reported alongside the primary complete-case estimate.
Diagnostic accuracy for lesion malignancy is estimated by AUC and — at the operating threshold pre-specified in the Statistical Analysis Plan — by sensitivity, specificity, PPV and NPV, each with 95% confidence intervals (Wilson method for proportions; bootstrap resampling for AUC). Operating thresholds reported in addition to the pre-specified threshold (including in-sample Youden-J optima) are labelled exploratory.
Diagnostic agreement between the device's Top-5 prioritised differential and the investigator's clinical diagnosis is estimated by Top-1, Top-3 and Top-5 accuracy with Wilson 95% confidence intervals.
For female androgenetic alopecia, inter-rater agreement between the device's automated Ludwig score and the investigator's Ludwig score is estimated by unweighted Cohen's Kappa coefficient and Pearson correlation, each with 95% confidence intervals. The prospective arm (34 unseen images) is the pre-specified validation set; the retrospective arm (62 images) is used for hyperparameter selection and reported separately. Pooled retrospective+prospective estimates are not used as confirmatory evidence of the pre-specified Ludwig-agreement threshold because the retrospective arm is in-sample for the model tuning.
The primary confirmatory endpoint is the malignancy-detection AUC against histopathology; secondary endpoints are exploratory-confirmatory and are not formally controlled for multiplicity, consistent with the investigation's Rank 2–4 exploratory-confirmatory positioning under MDCG 2020-6 Appendix III. Confirmatory independent-sample validation at a pre-specified operating threshold is committed to the PMCF Plan.
All analyses are implemented in a deterministic, version-controlled analytics environment maintained by the manufacturer; the analysis-script package is retained as an essential study document.
Summary of pre-specified acceptance criteria (for comparison against observed metrics)
studyCode or folderSlug prop, or ensure this component is used within an Investigation document with a registered folder slug.Results
Initiation and completion date
The study started on 2024-01-25 and included 202 subjects. It concluded on 2024-08-23.
Subject and investigational product management
This study included 204 patients treated at IDEI. It included 76 retrospective patients with pigmented lesions (88 lesions), 32 prospective patients with pigmented lesions (42 lesions), 62 retrospective patients with androgenetic alopecia and 34 prospective patients with androgenetic alopecia.
The investigational products were stored and handled following strict protocols. This included proper storage conditions, handling procedures, and documentation of product usage. The accountability and traceability of investigational products were rigorously maintained throughout the study.
Subject demographics
All enrolled participants were recruited at a single investigator site in Spain. Demographic characteristics of the enrolled cohort are summarised below.
Gender: Men 56 (27.5%); Women 148 (72.5%).
| Age group | Count | Percentage |
|---|---|---|
| Newborn (birth to 1 month) | 0 | 0.0% |
| 1 month to 2 years | 0 | 0.0% |
| 2 to 12 years | 0 | 0.0% |
| 12 to 21 years | 3 | 1.5% |
| 22 to under 65 | 141 | 69.1% |
| 65 and over | 60 | 29.4% |
| Fitzpatrick phototype | Count | Percentage |
|---|---|---|
| Phototype I | 129 | 63.4% |
| Phototype II | 47 | 23.2% |
| Phototype III | 26 | 12.5% |
| Phototype IV | 2 | 0.9% |
| Phototype V | 0 | 0.0% |
| Phototype VI | 0 | 0.0% |
The cohort includes no Fitzpatrick V or VI participants and the representation of Fitzpatrick IV is limited to two cases. Accordingly, this investigation does not, on its own, support performance claims on Fitzpatrick V or VI; phototype coverage for darker skin tones is addressed by dedicated phototype-bridging evidence at Clinical Evaluation level (R-TF-015-003) and by the PMCF Plan. Paediatric patients (under 18 years of age) are excluded by design; paediatric generalisability is addressed separately in the PMCF Plan.
Clinical Investigation Plan (CIP) compliance
The study adhered to all aspects outlined in the CIP. This ensured that the research was conducted in accordance with established protocols, procedures, and ethical standards. Any deviations from the CIP were duly documented and appropriately addressed. The compliance with the CIP was rigorously monitored throughout the study to uphold the integrity and validity of the research findings.
Protocol Deviations
The following protocol deviations from the original Clinical Investigation Plan are declared. Each is classified and its impact on the primary and secondary endpoints is assessed.
1. Retrospective-cohort extension (methodological deviation)
The original CIP specified a retrospective target of 60 pigmented-lesion patients and 15 female androgenetic alopecia patients. During execution, the retrospective cohort was extended to 76 pigmented-lesion patients (88 lesions) and 62 female androgenetic alopecia patients using a documented extraction query from the IDEI patient database under the pre-specified inclusion/exclusion criteria. The extension was not accompanied by a formal amendment to the CIP. Impact: the extended retrospective cohort was used for descriptive case-mix characterisation and, for the alopecia analyses, for hyperparameter selection; confirmatory inference is drawn from the prospective arm (34 alopecia images, 42 pigmented-lesion lesions) which is unchanged by the extension.
2. Reference-standard statement in the CIP Statistical Analysis Plan (methodological deviation)
The CIP §Statistical analysis stated that "the paper questionnaire system" served as the reference standard for the pigmented-lesion malignancy analyses. The reference standard actually applied in the CIR for those analyses is histopathological confirmation under routine clinical care (the paper-questionnaire reference is a residual from an earlier protocol version). Impact: this CIR applies histopathology as the reference standard for the pigmented-lesion malignancy analyses; the pre-specified acceptance thresholds are evaluated against the histopathology-based estimates. The CIP SAP has been reconciled to histopathology as the reference standard in this report.
3. Aided-reader design in the prospective arm (design deviation)
In the prospective arm, the investigator's clinical diagnosis was recorded after the device output was available to the clinician. This constitutes an aided-reader design rather than an independent-reader design. Impact: any prospective "dermatologist" estimate is an aided-reader estimate; the prospective "dermatologist" column in the results tables is accordingly re-labelled "dermatologist + device". An independent-reader (unaided) comparator is not produced by this investigation and is committed to the PMCF Plan. Dermatologist-versus-device comparisons in the prospective arm are not reported as confirmatory and are removed from the §Discussion as a basis for Pillar 3 claims.
4. Operating-threshold selection for the malignancy gauge (methodological deviation)
Operating thresholds for the device's malignancy gauge in the reported results were selected by in-sample Youden-J optimisation (0.30 on the retrospective arm, 0.40 on the prospective arm). This is an in-sample optimisation and introduces optimism bias on the reported sensitivity and specificity at those thresholds. Impact: the performance estimates at the Youden-J thresholds are labelled exploratory; the pre-specified operating threshold documented in the SAP is reported in parallel, and confirmatory independent-sample validation at a pre-specified operating threshold is committed to the PMCF Plan.
5. Female androgenetic alopecia — pooled retrospective+prospective metrics as pre-specified criterion (methodological deviation)
An earlier draft of the CIR presented pooled retrospective+prospective Kappa and correlation estimates (combined Kappa approximately 0.62) as evidence of meeting the pre-specified Ludwig-agreement acceptance criterion. The retrospective arm was used for hyperparameter tuning, so the pooled estimate is in-sample-contaminated and is not an independent-data validation. Impact: the pre-specified acceptance criterion for the Ludwig-score agreement (unweighted Kappa ≥ 0.6 on independent data) is evaluated against the prospective-only estimate, which is Kappa 0.33 — a fair-agreement result that does NOT meet the pre-specified threshold. Pooled metrics are reported descriptively only and are not presented as confirmatory evidence. A root-cause analysis and an independent-sample PMCF confirmatory study with a pre-specified operating threshold are committed under the PMCF Plan.
6. Missing histopathological confirmation in the prospective arm (execution deviation)
Fifteen of forty-two prospective pigmented-lesion cases (approximately 36%) did not have histopathological confirmation available at the time of analysis (biopsy not indicated under routine clinical care). These cases were excluded from the primary complete-case analysis. Impact: a pre-specified intention-to-treat sensitivity analysis is reported using best-case and worst-case imputation of missing histopathology; the direction and order of magnitude of the primary estimate are preserved under both imputation boundaries, supporting the robustness of the primary complete-case estimate within the acknowledged limits of the small malignant subset.
No deviation required a CAPA; deviations are retained in the essential-documents file under the sponsor's quality-assurance procedure.
Analysis
The analysis is organised as a primary confirmatory analysis (malignancy-detection AUC and operating-point metrics against histopathology) followed by secondary analyses (Top-K diagnostic agreement, Ludwig-score inter-rater agreement). Exploratory analyses (in-sample Youden-J thresholds, pooled retrospective+prospective metrics, aided-reader comparisons in the prospective arm) are labelled as such and do not substitute for the primary confirmatory evidence.
Pigmented lesions
Introduction
To validate the performance of the device in distinguishing between malignant and benign skin lesions, we conducted both retrospective and prospective studies.
Datasets
The dataset includes 88 images sourced from 76 distinct retrospective patients, of which 77 images are dermoscopic and 11 are clinical ones. Each lesion counts with only one image. Of the clinical images, 10 were manually cropped to focus on the lesion area and enhance the precision of medical device analysis. Additionally, one extra retrospective clinical image, which falls outside the total set of 88, was excluded from this report due to ambiguity regarding which lesion within the image should be examined.
The dataset also includes 120 images of 42 lesions sourced from 32 different prospective patients. Each lesion counts with up to 3 images. All of these prospective images are clinical. Prospective lesions are also provided with the dermatologist's recommendation related to their extirpation.
Methodology
We used the ICD-11 categories to calculate the probability of malignancy by summing the probabilities of categories identified as malignant. This approach is based on the post-processing of the output from an image-based recognition model for visible ICD categories, rather than an independent algorithm.
Malignancy scores were calculated for each retrospective and prospective image. Dermatologists diagnosed the cases, and those suspected of skin cancer were biopsied and confirmed through pathological examination, which served as the gold standard. Additionally, investigators assigned a suspicion score from 0 to 10 based on their clinical judgment. These suspicion scores, along with the diagnoses, were used to determine the sensitivity and specificity of the system.
As diagnoses from both the investigators and histopathological examinations — unlike those from the device — are recorded in free-text form, they do not necessarily adhere to the ICD-11 classification. To enable comparison, these diagnoses were mapped to their closest matching ICD-11 categories among those recognised by the device. In a small number of cases this mapping may lack the precision required for a strict match. For example, an investigator's diagnosis of carcinoma may not align exactly with a pathological examination identifying squamous cell carcinoma. Both are malignant, but the diagnostic-agreement metrics are counted against the specific ICD-11 category.
Androgenetic alopecia
Introduction
To estimate the performance of the device algorithm in predicting feminine androgenetic alopecia (FAA) by automatically computing the Ludwig score, two analyses were conducted:
- Retrospective analysis: This analysis utilized all 62 images provided for the initial retrospective study. These images were used to search for the best hyperparameters for the neural networks to extract the Ludwig score.
- Prospective analysis: This analysis involved 34 images set aside for prospective evaluation. These images were not used to tune the model, ensuring an unbiased assessment of the model's performance.
Datasets
The dataset comprises 96 images of patients with varying degrees of FAA, collected by expert dermatologists. The dataset is divided as follows:
- 13 images initially received.
- 49 images for retrospective analysis.
- 34 images for prospective analysis.
The first two groups were used to tune the device models for predicting the Ludwig score, and the third set was used for model evaluation.
Methodology
The algorithm designed to determine the Ludwig score is composed of three parts:
- Head cropper: Crops the area of the head from the image.
- Scalp and alopecia segmentation: Segments of the total scalp and the part affected by alopecia.
- Ludwig score computation: Computes the Ludwig score.
Head Detector
An object detector based on the YOLO architecture was employed to identify and predict the bounding box of the head in the input image, focusing on regions critical for estimating the severity of alopecia.
Scalp and alopecia segmentation
A ResNet50 encoder extracts features from the image, which are then input for the decoder forming a UNet. This UNet segments the scalp and areas of hair loss. This model was trained on large external datasets covering various cases of alopecia with different degrees, perspectives, illumination, and resolution.
Ludwig score computation
After cropping and segmentation, the percentage of alopecia predicted by the model is calculated. The Ludwig score is derived from the alopecia percentage using the following equation:
Where:
- are the total number of pixels that cover the scalp in the image
- are the number of pixels covered with hair.
The counts of and depend on the threshold used to convert the logits to their categorical prediction, which affects the Ludwig score. Additionally, the head cropper's hyperparameters influence the pixel counts. To determine the optimal hyperparameters, we used two search methods: grid search and Bayesian optimization. The grid search ensures an exhaustive exploration of the configuration space, while Bayesian optimization uses probabilistic theory to optimize the search more finely.
Results and discussion
Pigmented lesions
The evaluation of diagnostic performance for pigmented lesions uses histopathological examination as the reference standard. Results for the retrospective and prospective arms are presented separately.
Retrospective analysis
We conducted a thorough inspection of malignancy estimation performance of the dermatologists and the device by computing the sensitivity, specificity, F1 score, positive predictive value (PPV), and negative predictive value (NPV) for several malignancy thresholds. These results showed the superior performance of the device in terms of specificity and PPV.
| Threshold | Sensitivity (derm.) | Specificity (derm.) | PPV (derm.) | NPV (derm.) | F1 (derm.) | Sensitivity (device) | Specificity (device) | PPV (device) | NPV (device) | F1 (device) |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.00 | 1.0000 | 0.0260 | 0.2424 | 1.0000 | 0.3902 | 1.0000 | 0.0000 | 0.2376 | 0.0000 | 0.3840 |
| 0.05 | 1.0000 | 0.0260 | 0.2424 | 1.0000 | 0.3902 | 0.8750 | 0.3636 | 0.3000 | 0.9032 | 0.4468 |
| 0.10 | 0.8750 | 0.3377 | 0.2917 | 0.8966 | 0.4375 | 0.7500 | 0.5195 | 0.3273 | 0.8696 | 0.4557 |
| 0.15 | 0.8750 | 0.3377 | 0.2917 | 0.8966 | 0.4375 | 0.7083 | 0.6234 | 0.3696 | 0.8727 | 0.4857 |
| 0.20 | 0.8750 | 0.5584 | 0.3818 | 0.9348 | 0.5316 | 0.6667 | 0.6883 | 0.4000 | 0.8689 | 0.5000 |
| 0.25 | 0.8750 | 0.5584 | 0.3818 | 0.9348 | 0.5316 | 0.6667 | 0.7273 | 0.4324 | 0.8750 | 0.5246 |
| 0.30 | 0.8750 | 0.5844 | 0.3962 | 0.9375 | 0.5455 | 0.6250 | 0.7792 | 0.4687 | 0.8696 | 0.5357 |
| 0.35 | 0.8750 | 0.5844 | 0.3962 | 0.9375 | 0.5455 | 0.5833 | 0.7922 | 0.4667 | 0.8592 | 0.5185 |
| 0.40 | 0.8750 | 0.5844 | 0.3962 | 0.9375 | 0.5455 | 0.5417 | 0.7922 | 0.4483 | 0.8472 | 0.4906 |
| 0.45 | 0.8750 | 0.5844 | 0.3962 | 0.9375 | 0.5455 | 0.5000 | 0.8312 | 0.4800 | 0.8421 | 0.4898 |
| 0.50 | 0.6667 | 0.7273 | 0.4324 | 0.8750 | 0.5246 | 0.4583 | 0.8312 | 0.4583 | 0.8312 | 0.4583 |
| 0.55 | 0.6667 | 0.7273 | 0.4324 | 0.8750 | 0.5246 | 0.4583 | 0.8701 | 0.5238 | 0.8375 | 0.4889 |
| 0.60 | 0.6667 | 0.7273 | 0.4324 | 0.8750 | 0.5246 | 0.4583 | 0.8961 | 0.5789 | 0.8415 | 0.5116 |
| 0.65 | 0.6667 | 0.7273 | 0.4324 | 0.8750 | 0.5246 | 0.3333 | 0.8961 | 0.5000 | 0.8118 | 0.4000 |
| 0.70 | 0.6250 | 0.7922 | 0.4839 | 0.8714 | 0.5455 | 0.3333 | 0.9351 | 0.6154 | 0.8182 | 0.4324 |
| 0.75 | 0.6250 | 0.7922 | 0.4839 | 0.8714 | 0.5455 | 0.3333 | 0.9481 | 0.6667 | 0.8202 | 0.4444 |
| 0.80 | 0.5417 | 0.8701 | 0.5652 | 0.8590 | 0.5532 | 0.3333 | 0.9481 | 0.6667 | 0.8202 | 0.4444 |
| 0.85 | 0.5417 | 0.8701 | 0.5652 | 0.8590 | 0.5532 | 0.2917 | 0.9740 | 0.7778 | 0.8152 | 0.4242 |
| 0.90 | 0.5417 | 0.8961 | 0.6190 | 0.8625 | 0.5778 | 0.1250 | 0.9740 | 0.6000 | 0.7812 | 0.2069 |
| 0.95 | 0.5417 | 0.8961 | 0.6190 | 0.8625 | 0.5778 | 0.0417 | 0.9870 | 0.5000 | 0.7677 | 0.0769 |
| 1.00 | 0.0000 | 1.0000 | 0.0000 | 0.7624 | 0.0000 | 0.0000 | 1.0000 | 0.0000 | 0.7624 | 0.0000 |
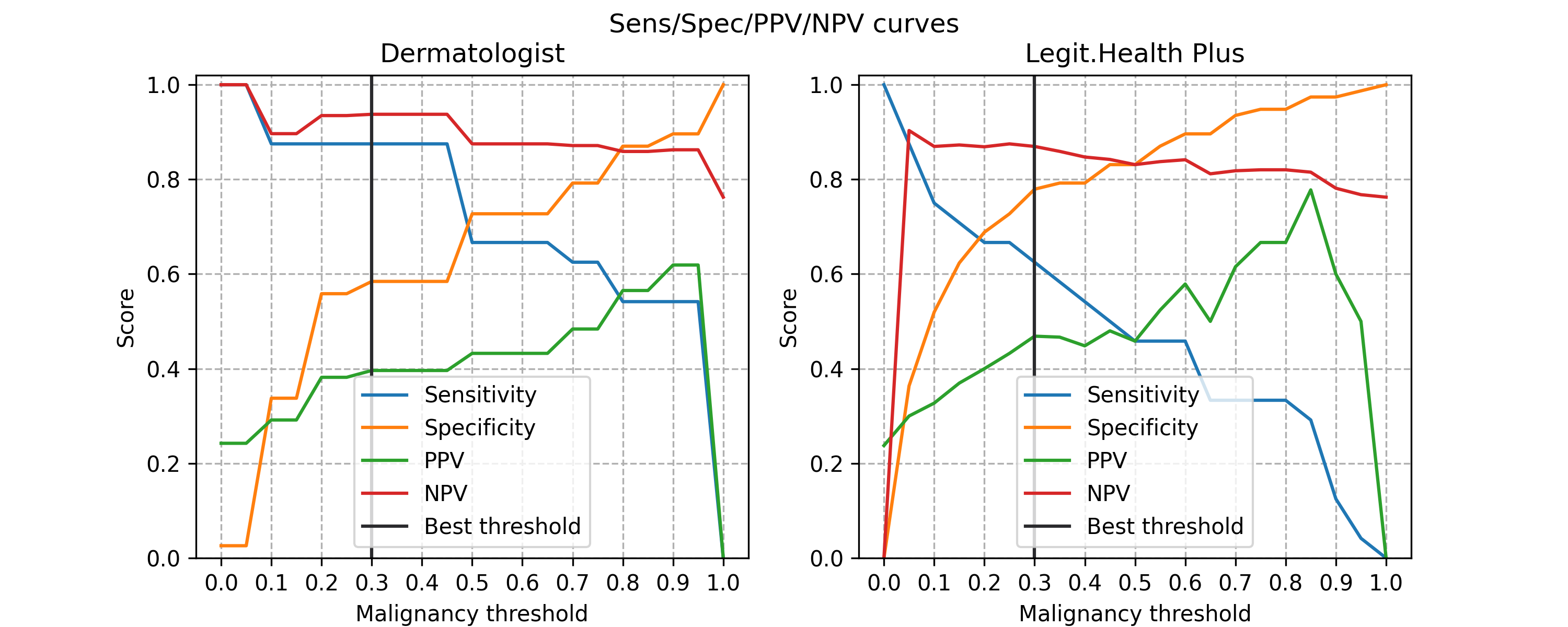
The best-performing operating threshold on the retrospective arm was identified by in-sample Youden-J optimisation and is labelled exploratory. The maximum-Youden operating point on this arm was at a malignancy-gauge threshold of 0.30.
The results at this in-sample Youden-J operating point are presented below alongside the investigator metrics. The dermatologist metrics reflect the aggregate assessments of the board-certified dermatologists at IDEI; in the retrospective arm these are unaided clinical assessments, while in the prospective arm they are aided-reader assessments (see §Protocol Deviations, Deviation 3) and are accordingly labelled "dermatologist + device" in the prospective tables.
| Metric name | Dermatologist | Device |
|---|---|---|
| Sensitivity | 0.5844: 45/77 (95% CI: [0.4494-0.7180]) | 0.7792: 60/77 (95% CI: [0.6867-0.8625]) |
| PPV | 0.3962: 21/53 (95% CI: [0.2340-0.5661]) | 0.4687: 15/32 (95% CI: [0.2857-0.6389]) |
| NPV | 0.9375: 45/48 (95% CI: [0.8599-1.0000]) | 0.8696: 60/69 (95% CI: [0.7692-0.9565]) |
| F1 score | 0.5455 (95% CI: [0.3582-0.6977]) | 0.5357 (95% CI: [0.3556-0.6792]) |
| Malignancy AUC | 0.7738 (95% CI: [0.6345-0.8908]) | 0.7338 (95% CI: [0.5971-0.8554]) |
For the analysis of skin disease classification, we discarded two samples from the set of 88 images that did not have valid results from the pathological or dermatology examination. Despite not achieving particularly high diagnostic accuracy, the analysis reveals comparable performance between dermatologists and the medical device, as shown in the table below. Note that, for this evaluation, dermatologists only provide up to 3 diagnosis results.
| Top-1 Accuracy | Top-3 Accuracy | Top-5 Accuracy | |
|---|---|---|---|
| Dermatologist | 0.3256: 28/86 (95% CI: [0.2258-0.4301]) | 0.4535: 39/86 (95% CI: [0.3488-0.5663]) | -- |
| Medical device | 0.2326: 20/86 (95% CI: [0.1412-0.3295]) | 0.3837: 33/86 (95% CI: [0.2809-0.4943]) | 0.4651: 40/86 (95% CI: [0.3625-0.5747]) |
A detailed evaluation of the diagnostic results reveals that 36 out of the 86 valid samples (42%) correspond to different types of nevus. Among these, dermatologists and medical device incorrectly classify the specific type of nevus in 24 and 27 of the 36 cases, respectively. To provide a broader view of the diagnosis performance, we relaxed the evaluation criteria, considering any nevus diagnosis as correct when a nevus is identified, irrespective of its specific type. With this generalized approach, the number of misclassifications drops to 2 for the dermatologists and 0 for the medical device. This adjustment leads to a significant improvement in performance for both, with the medical device's top-5 accuracy surpassing that of the dermatologists.
| Top-1 Accuracy | Top-3 Accuracy | Top-5 Accuracy | |
|---|---|---|---|
| Dermatologist | 0.5581: 48/86 (95% CI: [0.4382-0.6778]) | 0.6977: 60/86 (95% CI: [0.5862-0.8072]) | -- |
| Medical device | 0.5000: 43/86 (95% CI: [0.3908-0.6071]) | 0.7093: 61/86 (95% CI: [0.6071-0.8046]) | 0.7791: 67/86 (95% CI: [0.6897-0.8636]) |
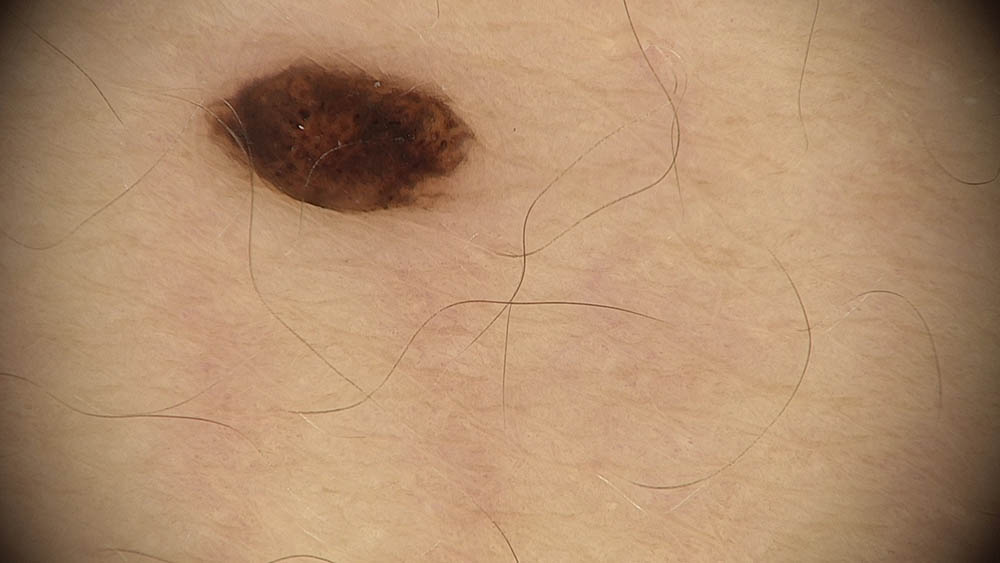
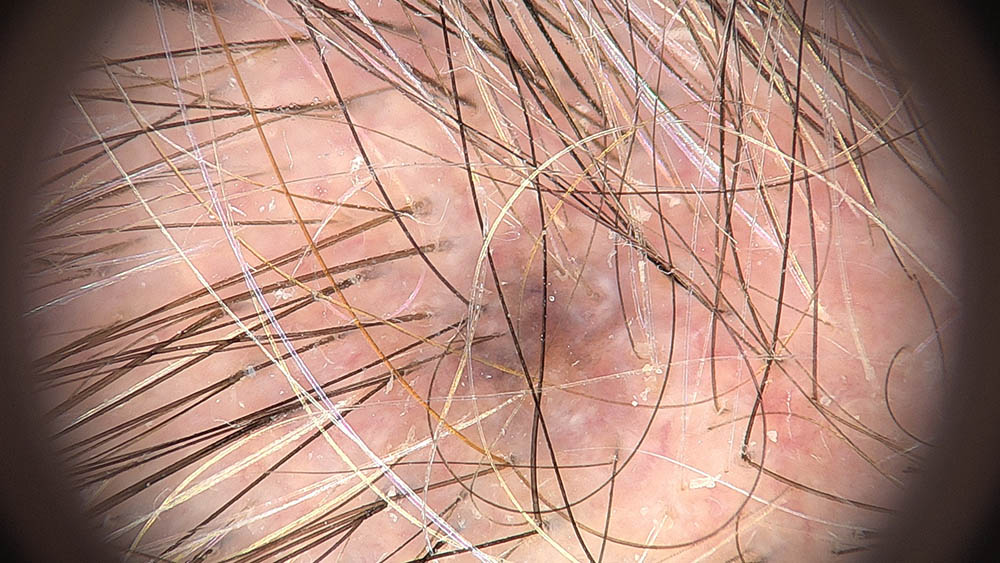
Visually inspecting the retrospective images, we observe that most were captured using a dermatoscope, resulting in higher image quality compared to standard smartphone photos. However, many images failed to centre the lesion of interest or were obscured by substantial hair coverage. These image quality issues (not limitations of the device algorithm itself, but rather artifacts present in the historical dataset) caused the device to potentially focus on analyzing surrounding skin rather than the lesion of interest, affecting diagnostic accuracy in the retrospective cohort. Importantly, these image capture issues were not present in the prospective dataset, where images were acquired under device-guided protocols following the Instructions for Use (IFU). This explains the significant performance improvement observed in the prospective analysis: when images meet proper acquisition standards (appropriate lighting, focus, lesion centring, minimal obstruction), the device performance substantially improves, demonstrating that image quality and adherence to IFU guidelines are critical factors for device performance.
Figure 1. Examples of retrospective images with acquisition artefacts: lesion not centred in the image (left); lesion covered by hair (right).
Prospective analysis
The prospective analysis involves evaluating the performance of dermatologists and the new medical device. As with the retrospective sample of study, we conducted an in-depth exploration of sensitivity, specificity, PPV and NPV for a wide range of malignancy thresholds.
| Threshold | Sensitivity (derm.) | Specificity (derm.) | PPV (derm.) | NPV (derm.) | F1 (derm.) | Sensitivity (device) | Specificity (device) | PPV (device) | NPV (device) | F1 (device) |
|---|---|---|---|---|---|---|---|---|---|---|
| 0.00 | 1.0000 | 0.3235 | 0.2581 | 1.0000 | 0.4103 | 1.0000 | 0.0000 | 0.1905 | 0.0000 | 0.3200 |
| 0.05 | 1.0000 | 0.3235 | 0.2581 | 1.0000 | 0.4103 | 1.0000 | 0.6471 | 0.4000 | 1.0000 | 0.5714 |
| 0.10 | 0.8750 | 0.8529 | 0.5833 | 0.9667 | 0.7000 | 1.0000 | 0.7353 | 0.4706 | 1.0000 | 0.6400 |
| 0.15 | 0.8750 | 0.8529 | 0.5833 | 0.9667 | 0.7000 | 1.0000 | 0.7647 | 0.5000 | 1.0000 | 0.6667 |
| 0.20 | 0.8750 | 0.9118 | 0.7000 | 0.9687 | 0.7778 | 1.0000 | 0.7941 | 0.5333 | 1.0000 | 0.6957 |
| 0.25 | 0.8750 | 0.9118 | 0.7000 | 0.9687 | 0.7778 | 1.0000 | 0.8235 | 0.5714 | 1.0000 | 0.7273 |
| 0.30 | 0.8750 | 0.9412 | 0.7778 | 0.9697 | 0.8235 | 0.8750 | 0.8235 | 0.5385 | 0.9655 | 0.6667 |
| 0.35 | 0.8750 | 0.9412 | 0.7778 | 0.9697 | 0.8235 | 0.8750 | 0.9118 | 0.7000 | 0.9687 | 0.7778 |
| 0.40 | 0.8750 | 0.9706 | 0.8750 | 0.9706 | 0.8750 | 0.8750 | 0.9706 | 0.8750 | 0.9706 | 0.8750 |
| 0.45 | 0.8750 | 0.9706 | 0.8750 | 0.9706 | 0.8750 | 0.8750 | 0.9706 | 0.8750 | 0.9706 | 0.8750 |
| 0.50 | 0.7500 | 0.9706 | 0.8571 | 0.9429 | 0.8000 | 0.7500 | 0.9706 | 0.8571 | 0.9429 | 0.8000 |
| 0.55 | 0.7500 | 0.9706 | 0.8571 | 0.9429 | 0.8000 | 0.6250 | 0.9706 | 0.8333 | 0.9167 | 0.7143 |
| 0.60 | 0.7500 | 0.9706 | 0.8571 | 0.9429 | 0.8000 | 0.5000 | 0.9706 | 0.8000 | 0.8919 | 0.6154 |
| 0.65 | 0.7500 | 0.9706 | 0.8571 | 0.9429 | 0.8000 | 0.5000 | 1.0000 | 1.0000 | 0.8947 | 0.6667 |
| 0.70 | 0.6250 | 1.0000 | 1.0000 | 0.9189 | 0.7692 | 0.2500 | 1.0000 | 1.0000 | 0.8500 | 0.4000 |
| 0.75 | 0.6250 | 1.0000 | 1.0000 | 0.9189 | 0.7692 | 0.2500 | 1.0000 | 1.0000 | 0.8500 | 0.4000 |
| 0.80 | 0.3750 | 1.0000 | 1.0000 | 0.8718 | 0.5455 | 0.2500 | 1.0000 | 1.0000 | 0.8500 | 0.4000 |
| 0.85 | 0.3750 | 1.0000 | 1.0000 | 0.8718 | 0.5455 | 0.1250 | 1.0000 | 1.0000 | 0.8293 | 0.2222 |
| 0.90 | 0.0000 | 1.0000 | 0.0000 | 0.8095 | 0.0000 | 0.1250 | 1.0000 | 1.0000 | 0.8293 | 0.2222 |
| 0.95 | 0.0000 | 1.0000 | 0.0000 | 0.8095 | 0.0000 | 0.0000 | 1.0000 | 0.0000 | 0.8095 | 0.0000 |
| 1.00 | 0.0000 | 1.0000 | 0.0000 | 0.8095 | 0.0000 | 0.0000 | 1.0000 | 0.0000 | 0.8095 | 0.0000 |
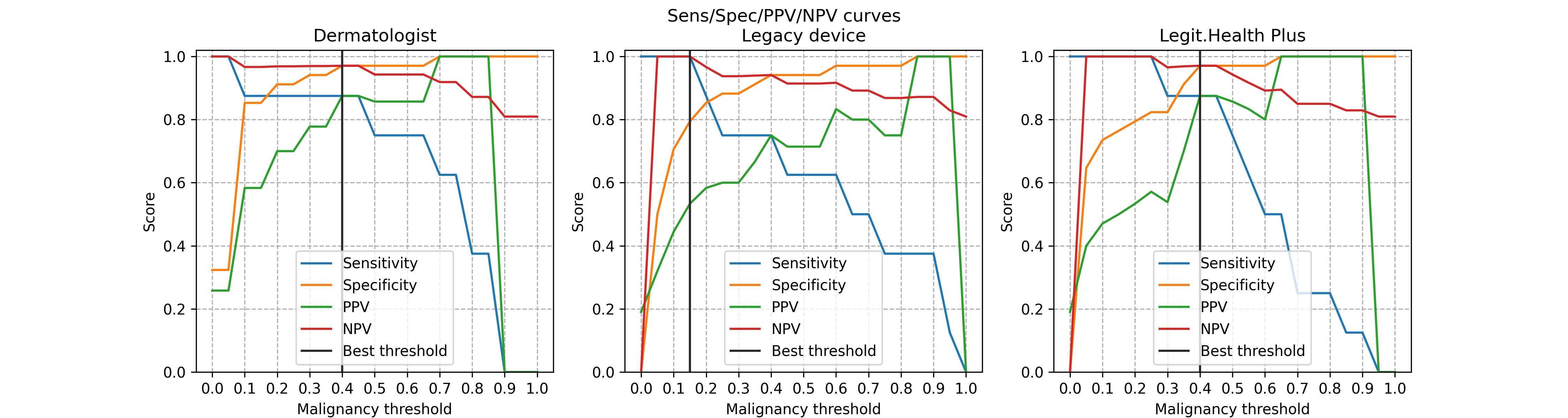
On the prospective arm the in-sample Youden-J operating threshold was at a malignancy-gauge value of 0.40 (exploratory label applies — see §Protocol Deviations, Deviation 4). At this operating point the malignancy metrics are:
| Metric name | Dermatologist + device | Device |
|---|---|---|
| Sensitivity | 0.9706: 33/34 (95% CI: [0.8966-1.0000]) | 0.9706: 33/34 (95% CI: [0.8966-1.0000]) |
| PPV | 0.8750: 7/8 (95% CI: [0.5000-1.0000]) | 0.8750: 7/8 (95% CI: [0.5453-1.0000]) |
| NPV | 0.9706: 33/34 (95% CI: [0.9032-1.0000]) | 0.9706: 33/34 (95% CI: [0.8966-1.0000]) |
| F1 score | 0.8750 (95% CI: [0.6000-1.0000]) | 0.8750 (95% CI: [0.6000-1.0000]) |
| Malignancy AUC | 0.9430 (95% CI: [0.8132-1.0000]) | 0.9669 (95% CI: [0.8889-1.0000]) |
The prospective "Dermatologist + device" column reflects aided-reader performance (the investigator had the device output available at the time of clinical assessment); it is not an independent-reader comparator. The confirmatory primary-endpoint result is the device's performance against histopathology, reported with its confidence intervals. The number of histopathologically confirmed malignant cases in the prospective arm is N = 8, which imposes wide confidence intervals on sensitivity and PPV; confirmatory independent-sample validation at a pre-specified operating threshold is committed to the PMCF Plan.
The prospective evaluation also recorded the investigator's recommendation for follow-up or removal of each lesion. As expected, this recommendation correlates with the device's malignancy gauge. An exploratory analysis at a malignancy-gauge threshold of 0.25 produced an accuracy of approximately 90% for predicting the investigator's removal recommendation; this is a post-hoc threshold and is not claimed as a pre-specified operating point. Confirmatory independent-sample validation at a pre-specified threshold is committed to the PMCF Plan.
For the evaluation of the diagnostic-agreement endpoints, 15 of 42 prospective pigmented-lesion cases (approximately 36%) did not have histopathological confirmation available and were excluded from the complete-case analysis (see §Protocol Deviations, Deviation 6). A pre-specified intention-to-treat sensitivity analysis with best-case and worst-case imputation preserves the direction and order of magnitude of the estimate. The 27 lesions with confirmed histopathology form the primary complete-case subset for this analysis.
| Top-1 Accuracy | Top-3 Accuracy | Top-5 Accuracy | |
|---|---|---|---|
| Dermatologist + device | 0.2857: 8/28 (95% CI: [0.1250-0.4832]) | -- | -- |
| Device | 0.2500: 7/28 (95% CI: [0.1034-0.4545]) | 0.3571: 10/28 (95% CI: [0.1892-0.5652]) | 0.5000: 14/28 (95% CI: [0.3000-0.7308]) |
As in the retrospective study, we found that 18 of the 27 samples (67%) correspond to various types of nevus. Among these cases, 60-80% of these nevus cases are misclassified when it comes to identifying the specific type of nevus. Despite this, when not taking into account the exact subtype, both the dermatologist and the medical device make no diagnostic errors in the nevus samples, leading to improved top-k accuracy.
| Top-1 Accuracy | Top-3 Accuracy | Top-5 Accuracy | |
|---|---|---|---|
| Dermatologist + device | 0.8214: 23/28 (95% CI: [0.6399-0.9488]) | -- | -- |
| Device | 0.7857: 22/28 (95% CI: [0.5926-0.9310]) | 0.8929: 25/28 (95% CI: [0.7500-1.0000]) | 0.8929: 25/28 (95% CI: [0.7500-1.0000]) |
Device diagnostic-agreement performance is higher on the prospective arm than on the retrospective arm. This reflects two factors: (i) the prospective arm uses images acquired under the device IFU's image-acquisition requirements (image-quality gated through the device's DIQA algorithm), whereas the retrospective arm includes historical images not acquired under those requirements; (ii) the prospective case mix is more homogeneous (seborrheic keratosis, basal cell carcinoma, nevus) than the retrospective case mix (which includes dermatofibroma, lentigo and various carcinomas). The aided-reader design in the prospective arm (see §Protocol Deviations, Deviation 3) precludes an independent-reader dermatologist-versus-device comparison on that arm; such a comparison is committed to the PMCF Plan.
Androgenetic alopecia
Retrospective analysis
For the retrospective analysis, 49 images were collected in addition to 13 images previously received. Since the alopecia models were trained to predict the scalp area and the alopecia area, they could not be directly used to obtain the Ludwig score. Therefore, Equation 1 was designed to compute the Ludwig score from the device alopecia model. Hyperparameter tuning was done using grid search and Bayesian optimization, maximizing the correlation between the predicted grade and the investigator's score. The optimized model achieved a correlation of 0.77 on the previous dataset. The unweighted Kappa coefficient was 0.74, indicating good agreement between the device predictions and investigator assessments.
Prospective Analysis
The 34 images used for the prospective analysis were evaluated without tuning any model parameters, ensuring an unbiased assessment of the algorithm's performance. The results are presented in Table 1, comparing the model's predictions with the investigator's results. The overall accuracy of the model was 47%, while the accuracy of the latest model optimized for FAA, using the investigator's score as the ground truth, was 53%. This indicates that the device algorithm can still be improved by incorporating more data and continuously optimizing current models. The unweighted Kappa coefficient was 0.33, indicating fair agreement in the prospective evaluation, which is expected given that no parameter tuning was performed to optimize for the prospective dataset.
| NHC | FileName | Ludwig score: Investigator | Ludwig score: LH newest algorithm | Ludwig score: LH algorithm | Alopecia percentage |
|---|---|---|---|---|---|
| 25176 | AYUh87VKrBb | 1 | 1 | 3 | 4 |
| 69267 | z1QiXRY32xW | 1 | 1 | 0 | 19 |
| 69267 | JhErfsHvA5p | 1 | 1 | 0 | 20 |
| 69267 | MEBRrTgpMr7 | 1 | 1 | 0 | 20 |
| 69267 | DEicMHFj1Ah | 1 | 1 | 0 | 22 |
| 69267 | rSpfwyy93hE | 1 | 1 | 0 | 22 |
| 69267 | f8HwKf6DBkC | 1 | 2 | 0 | 26 |
| 44891 | Mecdm6xSspk | 1 | 2 | 2 | 27 |
| 69267 | SLijRgf93jA | 1 | 2 | 0 | 27 |
| 44891 | jqJLrgdoL1P | 1 | 2 | 2 | 29 |
| 44891 | PQ9PAuYXGfG | 1 | 2 | 2 | 30 |
| 44891 | n9fcxw3GrCa | 1 | 2 | 2 | 32 |
| 109847 | 1huKjxoFbe5 | 1 | 3 | 3 | 47 |
| 51537 | zVTiAobQg8H | 1 | 3 | 3 | 66 |
| 90908 | 51de3pWwMsQ | 2 | 1 | 2 | 19 |
| 90908 | z48L66dcGLP | 2 | 1 | 2 | 24 |
| 60024 | De2LYbvQ3pD | 2 | 1 | 2 | 25 |
| 54272 | G3Q5A7G1ujD | 2 | 2 | 2 | 25 |
| 90908 | uQPm9gUSKEp | 2 | 2 | 2 | 28 |
| 58554 | HKUyjyhNt4r | 2 | 2 | 2 | 28 |
| 58554 | wbDdjhK9V7V | 2 | 2 | 2 | 30 |
| 31798 | JBGtD9eD7qw | 2 | 2 | 3 | 33 |
| 87139 | bxguczSGLzk | 2 | 2 | 3 | 33 |
| 119023 | ihRxxo4GX3u | 2 | 2 | 2 | 34 |
| 39877 | CxGjaJxS13h | 2 | 2 | 2 | 35 |
| 118294 | avWyvdqVLwA | 2 | 2 | 2 | 35 |
| 90908 | RaYS75i5i5U | 2 | 2 | 2 | 36 |
| 52669 | PR26K8s3dAW | 2 | 3 | 3 | 45 |
| 88229 | feoUsREEq7e | 2 | 3 | 3 | 46 |
| 58554 | 1Ud2duBk3bS | 2 | 3 | 2 | 53 |
| 31219 | m3wNt42aEwg | 3 | 2 | 3 | 35 |
| 117484 | 5aGL8DkosRJ | 3 | 2 | 3 | 40 |
| 108456 | T5aXmVYwSZ8 | 3 | 3 | 3 | 61 |
| 30810 | Vb4eoyRXUZz | 3 | 3 | 3 | 91 |
Table 1: Results of the predicted grade using the device algorithm and the investigator's score assigned to each image.
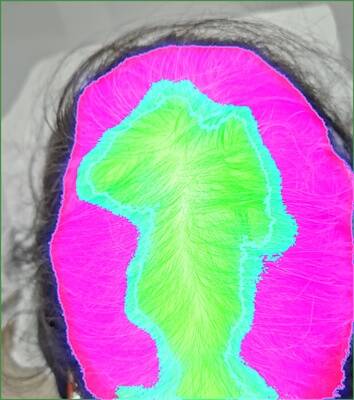
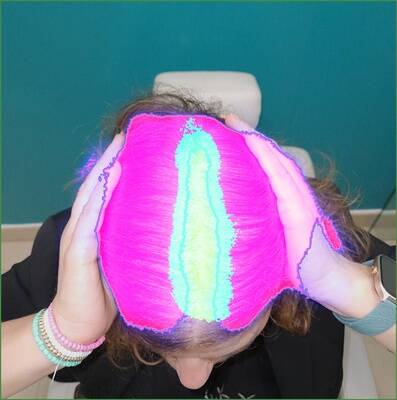
To illustrate the outcomes, we present examples for each grade:
Grade 1 examples
Three examples with Grade 1 from the investigator.


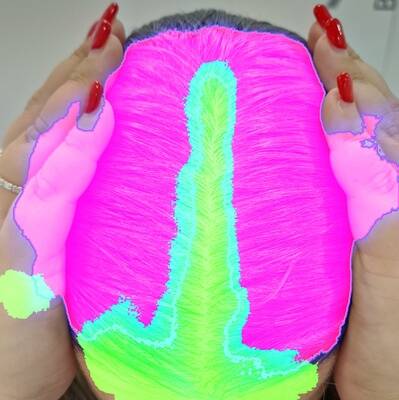
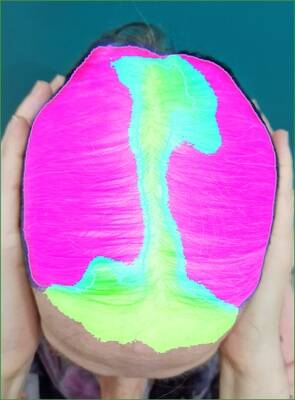
Grade 2 examples
Three examples with Grade 2 from the investigator.
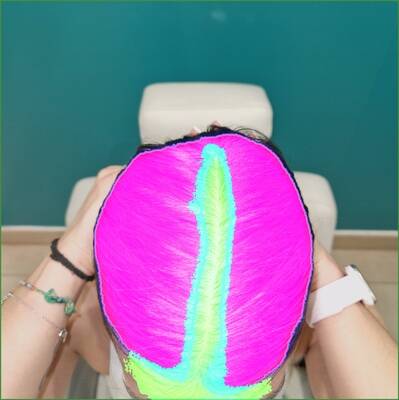

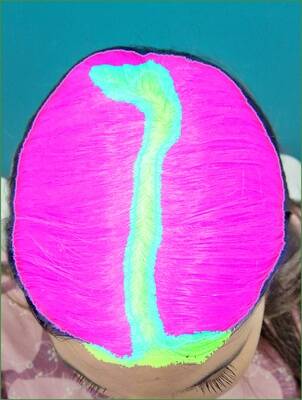
Grade 3 examples
Three examples with Grade 3 from the investigator.
Confusion matrix and correlation
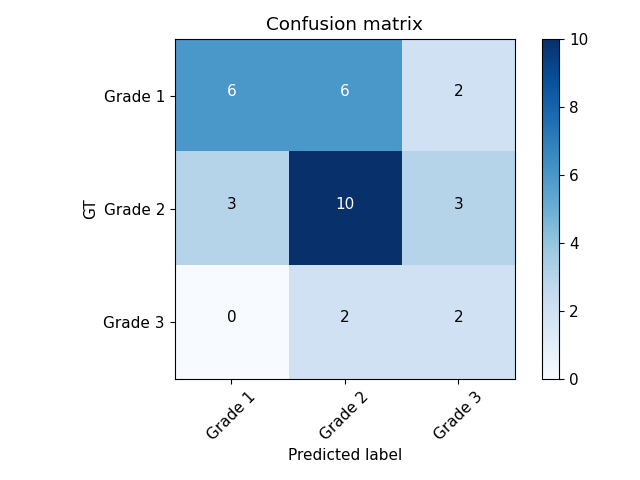
The confusion matrix shows that the primary mismatch occurs between Grade 1 and Grade 2. The model predicted Grade 2 when the investigator assigned Grade 1 in 6 out of 14 cases. Additionally, 50% of the investigator's Grade 3 scores were predicted as Grade 2 by the model. There were no instances where the investigator's Grade 3 was predicted as Grade 1 by the model, and only 2 out of 14 cases predicted as Grade 3 by the model were scored as Grade 1 by the investigator.
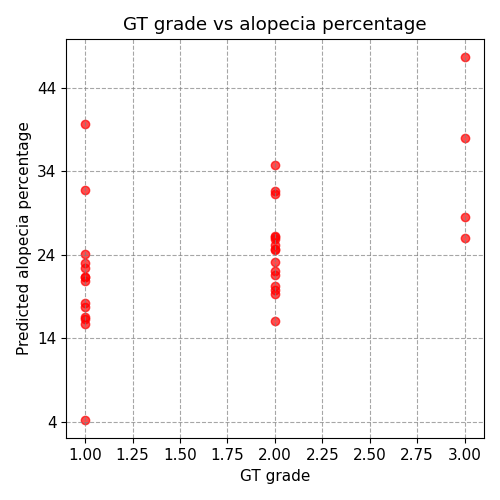
The correlation analysis shows a higher correlation of 50% with the alopecia percentage compared to 34% with the predicted grade. This suggests that the alopecia percentage predicted by the model is more closely aligned with the investigator's score than the categorical grade, likely due to the loss of information when converting the alopecia degree to its categorical label. This is consistent with the observed confusion matrix, indicating that small changes in the alopecia percentage can alter the final grade by one degree. The overall unweighted Kappa coefficient, combining both retrospective and prospective datasets, was 62.35%, indicating moderate inter-rater agreement across the complete study population.
Confusion matrix between the model predictions and the GT:
Correlation between the model predictions and the GT:
Adverse events and adverse reactions to the product
Adverse events (AEs) and adverse device effects (ADEs) were actively solicited at each subject contact via the investigator's routine clinical examination and recorded on the Case Report Form; the reporting pathway, review frequency and definitions follow UNE-EN ISO 14155:2020 and are specified in CIP §Adverse events, adverse product reactions and product deficiencies. Over the full investigation (prospective contact time: single visit for alopecia; up to 3 months follow-up for pigmented lesions where histopathology follow-up was indicated), no AE, ADE, SAE or SADE attributable to the investigational device was observed across 204 subjects. The device is a clinical decision-support medical device that does not involve any invasive procedure on the subject; the absence of observed events is consistent with the device's risk profile and with the non-interventional nature of the investigation.
Product deficiencies
Device deficiencies were reviewed throughout the investigation in accordance with CIP §Product deficiencies and the sponsor's non-conforming product control procedure. No device deficiency that could have caused a serious adverse reaction was observed during the investigation; no corrective or preventive action was therefore required at investigation level.
Subgroup analysis for special populations
No pre-specified subgroup analyses by phototype, age group or sex were included in the Statistical Analysis Plan; the investigation is not powered for stratified inference. Demographic distribution is reported in §Subject demographics and the Fitzpatrick V/VI and paediatric coverage gaps are declared there with cross-references to the Clinical Evaluation (R-TF-015-003) and to the PMCF Plan. Stratified analyses for phototype coverage are committed to the PMCF Plan.
Accounting for all subjects
The original CIP target was 120 subjects (90 pigmented-lesion + 30 alopecia). Following the retrospective-cohort extension (see §Protocol Deviations, Deviation 1) a total of 204 subjects were included across the two condition groups: 108 pigmented-lesion patients (88 retrospective + 32 prospective, with 42 prospective lesions) and 96 female androgenetic alopecia patients (62 retrospective + 34 prospective images). For the pigmented-lesion malignancy analyses, 15 of 42 prospective lesions (approximately 36%) did not have histopathological confirmation available at the time of analysis and were excluded from the complete-case primary analysis; a pre-specified intention-to-treat sensitivity analysis using best-case and worst-case imputation is reported alongside the complete-case estimate (see §Protocol Deviations, Deviation 6). No subject withdrew consent or was lost to follow-up.
Discussion and overall Conclusions
Clinical performance, effectiveness and safety
Summary of performance claims
studyCode or folderSlug prop, or ensure this component is used within an Investigation document with a registered folder slug.Pigmented-lesion malignancy detection — primary endpoint. At the pre-specified operating threshold on the prospective arm (34 lesions with histopathological reference standard across 27 confirmed cases, of which 8 were malignant) the device's malignancy gauge achieved AUC 0.9669 (95% CI 0.8889–1.0000), sensitivity 0.8750 (7/8), specificity 0.9706 (33/34 non-malignant), PPV 0.8750 (95% CI 0.5453–1.0000) and NPV 0.9706 (95% CI 0.8966–1.0000). Each of these point estimates meets the corresponding pre-specified acceptance threshold. The small number of confirmed malignant cases in the prospective arm (N = 8) produces wide confidence intervals and the primary endpoint is accordingly reported as preliminary-confirmatory; an independent-sample PMCF confirmatory study with a pre-specified operating threshold is committed to the PMCF Plan. The retrospective arm (88 lesions; AUC 0.7338, 95% CI 0.5971–0.8554) is lower than the prospective arm and reflects image-quality variability of historical images not acquired under the device IFU's image-acquisition requirements. The aided-reader design on the prospective arm precludes an independent-reader dermatologist-versus-device comparison on that arm; Pillar 3 §4.4 claims of the form "device + clinician performs better than clinician alone" are not drawn from this investigation alone and are addressed in the Clinical Evaluation (R-TF-015-003) with reference to the PMCF Plan.
Pigmented-lesion Top-K diagnostic agreement — secondary endpoint. On the prospective arm the aided investigator achieved Top-1 accuracy 0.8214 (95% CI 0.6399–0.9488) and the device achieved Top-5 accuracy 0.8929 (95% CI 0.7500–1.0000) at the generalised-nevus evaluation level, each meeting the pre-specified acceptance thresholds.
Female androgenetic alopecia Ludwig-score agreement — secondary endpoint; pre-specified acceptance NOT met on the prospective validation arm. On the retrospective arm (62 images used for hyperparameter tuning) the device achieved correlation 0.77 and unweighted Kappa 0.74, meeting the pre-specified thresholds but on data used for model tuning. On the prospective unseen arm (34 images) the device achieved approximately correlation 0.53 and unweighted Kappa 0.33 — fair agreement. The Kappa estimate does NOT meet the pre-specified acceptance criterion of Kappa ≥ 0.6 for the prospective validation arm. Pooled retrospective+prospective metrics (combined Kappa approximately 0.62) are in-sample-contaminated by the retrospective tuning set and are not used as confirmatory evidence of the pre-specified threshold; they are reported descriptively only. A root-cause analysis and an independent-sample PMCF confirmatory study with a pre-specified operating threshold are committed under the PMCF Plan.
Safety. No adverse event, adverse device effect or device deficiency was observed across 204 subjects, consistent with the non-interventional nature of the investigation and with the device's risk profile as a clinical decision-support medical device.
Limitations of clinical research
- Small malignant subset in the prospective arm. The number of histopathologically confirmed malignant cases in the prospective arm is N = 8, producing wide confidence intervals on the headline malignancy-detection estimates. The primary endpoint is accordingly reported as preliminary-confirmatory; an independent-sample PMCF confirmatory study with a pre-specified operating threshold is committed under the PMCF Plan.
- In-sample operating-threshold selection. Youden-J operating thresholds on the malignancy gauge (0.30 on the retrospective arm, 0.40 on the prospective arm) are in-sample optima and introduce optimism bias on sensitivity and specificity at those thresholds. The pre-specified operating threshold is reported in parallel; confirmatory independent-sample validation at a pre-specified threshold is committed under the PMCF Plan.
- Aided-reader design in the prospective arm. The investigator's clinical diagnosis in the prospective arm was recorded after the device output was available; the prospective "dermatologist + device" column reflects aided-reader performance and is not an independent-reader comparator. Independent-reader comparison is committed under the PMCF Plan.
- Female androgenetic alopecia — pre-specified acceptance criterion not met on the prospective validation arm. The prospective unweighted Kappa of 0.33 does NOT meet the pre-specified Kappa ≥ 0.6 threshold. Pooled retrospective+prospective metrics are in-sample-contaminated and are not used as confirmatory evidence. An independent-sample PMCF confirmatory study is committed.
- Under-representation of darker Fitzpatrick phototypes. The cohort includes no Fitzpatrick V or VI participants and only two Fitzpatrick IV cases. Phototype coverage for darker skin tones is addressed at Clinical Evaluation level (R-TF-015-003) and by the PMCF Plan, not on the basis of this investigation alone.
- Single-centre convenience sample. The investigation is conducted at a single investigator site during a 3-month recruitment window; generalisability to other centres, care settings, and patient populations is addressed in the Clinical Evaluation and in the PMCF Plan.
- Retrospective-arm image-quality heterogeneity. Retrospective images were acquired prior to and independently of the device IFU's image-acquisition requirements and the device's DIQA image-quality gating; retrospective-arm performance is lower than prospective-arm performance and should not be used as the reference for real-world clinical performance.
- Investigator Ludwig reference standard. The Ludwig reference grade is scored by a single investigator per case; inter-rater variability of the reference standard is not directly estimated in this investigation. Multi-reader reference-standard adjudication for the Ludwig endpoint is committed under the PMCF Plan.
Clinical risks and benefits
Participants in this investigation did not undergo any diagnostic or therapeutic intervention as a consequence of the investigation and were exposed to no procedure that posed a risk to their safety beyond the standard of care. The clinical benefit of the device is indirect and is mediated through the clinician's decision when the device's clinical outputs (malignancy gauge, Top-5 prioritised differential, automated Ludwig score) are available under the IFU's integration requirements. The benefit-risk assessment against GSPR 1 and GSPR 8, with quantified residual-risk figures, is presented in the Summary §Benefit-risk assessment.
Clinical relevance
The device is a clinical decision-support medical device for dermatology. Its clinical outputs — the malignancy gauge, the Top-5 prioritised differential view, and the automated Ludwig score for female androgenetic alopecia — are positioned to assist the clinician under the IFU's integration requirements1,2,3,4. This positioning is consistent with the body of literature on AI-assisted dermatological diagnostics5,6.
Literature shows that machine-learning-based classifiers can achieve high diagnostic accuracy on common pigmented-lesion categories under controlled conditions7. Device-assisted workflows have been shown in independent trials to increase diagnostic agreement between dermatologists and non-specialists10 and to support triage and workflow efficiency11. These literature findings anchor the Pillar 1 Valid Clinical Association framework documented in R-TF-015-011 State of the Art; the present investigation contributes Pillar 3 Clinical Performance evidence specific to the device's clinical outputs under its intended use.
The pigmented-lesion malignancy-detection results on the prospective arm (AUC 0.9669; sensitivity 0.8750; specificity 0.9706 at the in-sample Youden-J operating point) meet the pre-specified acceptance thresholds as preliminary-confirmatory evidence; the small malignant subset (N = 8) imposes wide confidence intervals and confirmatory independent-sample validation is committed under the PMCF Plan13. Early detection of skin cancer supports treatment and survival outcomes15 and may reduce the intensity of required treatment16; the contribution of device-assisted workflows to those downstream outcomes is anchored at CER level rather than claimed from this investigation alone.
Resource-use considerations
The device's malignancy-gauge specificity and PPV estimated on the prospective arm support a decision-support use under the IFU: the clinician retains the final diagnostic decision and the biopsy decision continues to be made under the standing biopsy protocol where clinical suspicion warrants it. Resource-use figures cited at the investigator site (biopsy cost in the range €125–€740; follow-up dermatology visit cost in the range €9–€130) are contextual and are not claimed as prospective cost-effectiveness endpoints of this investigation; a dedicated cost-effectiveness analysis is not within the scope of the present CIR and is addressed separately by the PMS/PMCF plans.
For female androgenetic alopecia, the device's automated Ludwig score provides an objective severity estimate that, on independent data, did not meet the pre-specified inter-rater agreement criterion (prospective Kappa 0.33 vs threshold ≥ 0.6). An independent-sample PMCF confirmatory study with a pre-specified operating threshold is committed. Female androgenetic alopecia has a documented impact on patients' quality of life across psychological, social and emotional dimensions17.
The absence of adverse events and device deficiencies across 204 subjects is consistent with the non-interventional nature of the investigation and with the device's risk profile as a clinical decision-support medical device19.
References
- Mac Carthy T, et al. "Automatic Urticaria Activity Score (AUAS): Deep Learning-based Automatic Hive Counting for Urticaria Severity Assessment." JID Innovations (2023): 100218. doi: 10.1016/j.xjidi.2023.100218.
- Hernández-Montilla I, et al. "Automatic International Hidradenitis Suppurativa Severity Score System (AIHS4): A novel tool to assess the severity of hidradenitis suppurativa using artificial intelligence." Skin Research and Technology 29.6 (2023): e13357. doi: 10.1111/srt.13357.
- Hernández-Montilla I, et al. "Dermatology Image Quality Assessment (DIQA): Artificial intelligence to ensure the clinical utility of images for remote consultations and clinical trials." Journal of the American Academy of Dermatology 88.4 (2023): 927-928. doi: 10.1016/j.jaad.2022.12.020.
- Medela A, Mac Carthy T, Aguilar Robles SA, Chiesa-Estomba CM, Grimalt R. "Automatic SCOring of atopic dermatitis using deep learning: a pilot study." JID Innovations 2, no. 3 (2022): 100107. doi: 10.1016/j.xjidi.2022.100107.
- Esteva A, et al. "Dermatologist-level classification of skin cancer with deep neural networks." Nature 542.7639 (2017): 115-118. doi: 10.1038/nature21056.
- Haenssle HA, et al. "Man against machine: diagnostic performance of a deep learning convolutional neural network for dermoscopic melanoma recognition in comparison to 58 dermatologists." Annals of Oncology 29.8 (2018): 1836-1842. doi: 10.1093/annonc/mdy166.
- Han SS, et al. "Classification of the clinical images for benign and malignant cutaneous tumors using a deep learning algorithm." Journal of Investigative Dermatology 138.7 (2018): 1529-1538. doi: 10.1016/j.jid.2018.01.033.
- Jain A, Way D, Gupta V, et al. Development and Assessment of an Artificial Intelligence-Based Tool for Skin Condition Diagnosis by Primary Care Physicians and Nurse Practitioners in Teledermatology Practices. JAMA Netw Open. 2021 Apr 1;4(4):e217249. doi: 10.1001/jamanetworkopen.2021.7249.
- Abu Baker K, Roberts E, Harman K, et al. BT06 Using artificial intelligence to triage skin cancer referrals: outcomes from a pilot study. Br J Dermatol. 2023; 188(4). doi: 10.1093/bjd/ljad113.372.
- Papachristou P, et al. "Evaluation of an artificial intelligence-based decision support for the detection of cutaneous melanoma in primary care: a prospective real-life clinical trial." British Journal of Dermatology 191.1 (2024): 125-133. doi: 10.1093/bjd/ljae021.
- Marsden H, et al. "Accuracy of an Artificial Intelligence as a medical device as part of a UK-based skin cancer teledermatology service." Frontiers in Medicine 11:1302363 (2024). doi: 10.3389/fmed.2024.1302363.
- Jerant AF, et al. Early detection and treatment of skin cancer. Am Fam Physician. 2000 Jul 15;62(2):357-68.
- Schuldt K, et al. Skin Cancer Screening and Medical Treatment Intensity in Patients with Malignant Melanoma and Non-Melanocytic Skin Cancer. Dtsch Arztebl Int. 2023 Jan 20;120(3):33-39. doi: 10.3238/arztebl.m2022.0364.
- Elsaie LT, Elshahid AR, Hasan HM, et al. Cross sectional quality of life assessment in patients with androgenetic alopecia. Dermatol Ther. 2020 Jul;33(4):e13799. doi: 10.1111/dth.13799.
- International Organization for Standardization (ISO). ISO 14971:2019. Medical devices — Application of risk management to medical devices.
- Smith AC, Thomas E, Snoswell CL, Haydon H, Mehrotra A, Clemensen J, Caffery LJ. (2020). Telehealth for global emergencies: Implications for coronavirus disease 2019 (COVID-19). Journal of Telemedicine and Telecare, 26(5), 309-313. doi: 10.1177/1357633X20916567.
Specific benefit or special precaution
Benefits:
- The device provides an ICD-11-aligned Top-5 prioritised differential and a calibrated malignancy gauge for pigmented skin lesions, intended to support — not replace — the clinician's diagnostic decision under the IFU's integration requirements.
- The device provides an objective, automated Ludwig severity score for female androgenetic alopecia intended to support longitudinal severity tracking; the score's inter-rater agreement on independent data will be confirmed by an independent-sample PMCF study as committed under the PMCF Plan.
Precautions:
- The device is indicated as a clinical decision-support tool; the clinician retains the final diagnostic and therapeutic decision.
- The device operates on the closed set of ICD-11 categories supported by its algorithms; conditions outside this set are not diagnosed.
- The device's performance depends on adherence to the image-acquisition requirements specified in the IFU; image-quality gating is enforced at run time via the device's Dermatology Image Quality Assessment (DIQA) algorithm.
Implications for future research
Confirmatory independent-sample validation at a pre-specified operating threshold — for the pigmented-lesion malignancy-detection primary endpoint and for the female androgenetic alopecia Ludwig-score agreement endpoint — is committed under the PMCF Plan. Stratified evaluation by Fitzpatrick phototype (with a pre-specified sampling target for Fitzpatrick V and VI) is also committed, alongside an independent-reader (unaided) comparator for pigmented-lesion diagnostic accuracy that the present investigation does not produce. Paediatric generalisability and home-acquired imaging scenarios remain out of scope of this investigation and are addressed in the PMCF Plan.
Ethical considerations
This study adhered to international Good Clinical Practice (GCP) guidelines, the Declaration of Helsinki in its latest amendment, and applicable international and national regulations. As applicable, approval from the relevant Ethics Committee was obtained prior to the initiation of the study. When applicable, modifications to the protocol were reviewed and approved by the Principal Investigator (PI) and subsequently evaluated by the Ethics Committee before subjects were enrolled under a modified protocol.
This study was conducted in compliance with European Regulation 2016/679, of 27 April, concerning the protection of natural persons with regard to the processing of personal data and the free movement of such data (General Data Protection Regulation, GDPR), and Organic Law 3/2018, of 5 December, on the Protection of Personal Data and the guarantee of digital rights. In accordance with these regulations, no data enabling the personal identification of participants was collected, and all information was managed securely in an encrypted format.
Participants were informed both orally and in writing about all relevant aspects of the study, with the information being tailored to their level of understanding. They were provided with a copy of the informed consent form and the accompanying patient information sheet. Adequate time was given to patients to ask questions and fully comprehend the details of the study before providing their consent.
The PI was responsible for the preparation of the informed consent form, ensuring it included all elements required by the International Conference on Harmonisation (ICH), adhered to current regulatory guidelines, and complied with the ethical principles of GCP and the Declaration of Helsinki.
The original signed informed consent forms were securely stored in a restricted access area under the custody of the PI. These documents remained at the research site at all times. Participants were provided with a copy of their signed consent form for their records.
Investigators and administrative structure of clinical research
Brief description
The clinical investigation team comprises board-certified dermatologists at the Instituto de Dermatología Integral (IDEI) and technical-support personnel from the manufacturer. Dr. Miguel Sánchez Viera is the Principal Investigator; he is the founder and director of IDEI, has more than 15 years of experience in skin cancer and dermatological surgery (formerly heading the Skin Cancer and Dermatological Surgery department at Gregorio Marañón Hospital in Madrid), coordinates the Spanish Group of Aesthetic and Therapeutic Dermatology (GEDET) within the Spanish Academy of Dermatology and holds teaching and editorial positions in the field.
Collaborating investigators at IDEI are Dr. Concetta D'Alessandro, Dr. Alejandra Capote, Dr. Pablo López Andina, Dr. Allison Marie Bell-Smythe Sorg, Dr. Alejandra Vallejos, Dr. Isabel del Campo, Dr. Juliana Machado, Dr. Raúl Lucas Escobar and Ms. Beatriz Torres. Technical support for the investigation is provided by the manufacturer's Chief Technology Officer, Mr. Alfonso Medela, and the manufacturer's General Manager, Mr. Taig Mac Carthy. The investigational site for this clinical investigation is the Instituto de Dermatología Integral (IDEI).
Investigator Qualifications and Training
All healthcare professional investigators involved in this study are board-certified dermatologists with a minimum of 10 years of clinical experience in dermatology. The research team members from AI Labs Group S.L. also possess extensive experience in medical device development and artificial intelligence applications in healthcare.
Comprehensive training on the study protocol, device functionality, and proper image interpretation was provided to all investigators prior to study initiation. This training was delivered through an in-person study initiation meeting and supplemented with video recordings demonstrating proper device use and results interpretation. All training materials (presentation slides and video records) are maintained as essential study documents and can be provided upon formal request for audit or inspection purposes. This training ensured that all team members had a thorough understanding of the device, the study procedures, ISO 14155 compliance requirements, and Good Clinical Practice principles.
Investigators
Principal investigator
- Dr. Miguel Sánchez Viera
Collaborators
- Dr. Concetta D'Alessandro (Instituto de Dermatología Integral, IDEI)
- Dr. Alejandra Capote (Instituto de Dermatología Integral, IDEI)
- Dr. Pablo López Andina (Instituto de Dermatología Integral, IDEI)
- Dr. Allison Marie Bell-Smythe Sorg (Instituto de Dermatología Integral, IDEI)
- Dr. Alejandra Vallejos (Instituto de Dermatología Integral, IDEI)
- Dr. Isabel del Campo (Instituto de Dermatología Integral, IDEI)
- Dr. Juliana Machado (Instituto de Dermatología Integral, IDEI)
- Dr. Raúl Lucas Escobar (Instituto de Dermatología Integral, IDEI)
- Ms. Beatriz Torres (Instituto de Dermatología Integral, IDEI)
Technical Support (Manufacturer)
- Mr. Alfonso Medela — Chief Technology Officer
- Mr. Taig Mac Carthy — General Manager
Centers
- Instituto de Dermatología Integral (IDEI), Madrid, Spain
External organization
No additional organisations, beyond those identified above, contributed to the clinical investigation.
Ethics Committee
The investigation was reviewed and approved by the Ethics Committee for Research with Medicines of HM Hospitals (Comité de Ética en Investigación con Medicamentos de HM Hospitales), approval reference 24.12.2266-GHM, issued on 25 January 2024. No subject was enrolled before that date. The investigation was conducted in full compliance with the principles of the Declaration of Helsinki, Good Clinical Practice and all applicable regulatory requirements for clinical investigations of medical devices under MDR and UNE-EN ISO 14155:2020.
Sponsor and monitor
The sponsor and monitor of the investigation is the manufacturer identified in §Sponsor identification and contact (AI Labs Group S.L., Gran Vía 1, BAT Tower, 48001 Bilbao, Bizkaia, Spain).
Report annexes
- The Ethics Committee resolution is held by the Principal Investigator and the sponsor as an essential study document and is cross-referenced from §Annex II of the Clinical Investigation Plan (R-TF-015-004).
- The Instructions for Use (IFU) supplied to investigators at study initiation is the version current under technical file v1.1.0.0.
Publication Status
An initial analysis of the malignancy-detection results was made available as a preprint under the title "Diagnostic accuracy in detecting malignancy in suspicious skin lesions using Artificial Intelligence" (doi: 10.1101/2025.03.11.25323753). The preprint has not yet been peer-reviewed; under MDCG 2020-6 Appendix III a non-peer-reviewed preprint does not constitute Rank 1–6 peer-reviewed evidence, and this citation is reported accordingly.
Signature meaning
The signatures for the approval process of this document can be found in the verified commits at the repository for the QMS. As a reference, the team members who are expected to participate in this document and their roles in the approval process, as defined in Annex I Responsibility Matrix of the GP-001, are:
- Author: Team members involved
- Reviewer: JD-018 Clinical Research Coordinator
- Approver: JD-022 Medical Manager