Declaración UE de Conformidad (Reglamento (UE) 2017/745)
Justificación técnica y legal de la conformidad de Legit.Health con el MDR (Clase IIb)
Table of contents
- Objetivo
- Marco normativo aplicable
- Conformidad con el MDR
- Exclusión no justificada
- Anexos
- Anexo I: Registro de Dispositivo Sanitario en EUDAMED
- Anexo II: Licencia de fabricación de producto sanitario emitida por la AEMPS
- Anexo III: Certificado de mantener un Sistema de Gestión de la Calidad de acuerdo a la norma ISO 13485
- Anexo IV: Contrato con un Organismo Notificado con fecha previa al 26 de mayo de 2024
- Anexo V: Declaración de Conformidad con MDD, incluyendo aviso de que el dispositivo no ha experimentado cambios significativos
- Anexo VI: Carta del Organismo Notificado confirmando el estado de solicitud formal en relación a la disposición transitoria del MDR
- Declaración de Conformidad y marcado CE
Objetivo
Este documento tiene como objetivo demostrar, tanto desde el punto de vista técnico como legal, que el software de dispositivo sanitario Legit.Health, desarrollado por AI Labs Group SL, cumple con los requisitos del Reglamento (UE) 2017/745 (MDR) para la clase de riesgo IIb, gracias a su certificación previa en virtud de la Directiva 93/42/EEC (MDD), y que puede ser utilizado en el marco de la licitación pública AQUAS-2025-42 sin ningún riesgo de incumplimiento normativo y ningún motivo de rechazo justificado.
Marco normativo aplicable
Como dispositivo sanitario, aplican las siguientes normativas:
- Reglamento (UE) 2017/745 (MDR): aplicable desde el 26 de mayo de 2020, con un período transitorio que permite la continuidad de los dispositivos MDD hasta el 27 de mayo de 2025, siempre que cumplan ciertos requisitos (Art. 120).
- Directiva 93/42/EEC (MDD): aplicable hasta el 26 de mayo de 2020.
Conformidad con el MDR
La licitación AQUAS-2025-42 establece como requisito la conformidad del producto sanitario con el Reglamento (UE) 2017/745, en adelante, “MDR”. Este reglamento es el reglamento europeo de dispositivos sanitarios, que reemplaza al anterior Directiva 93/42/CEE, en adelante “MDD”.
La conformidad con el MDR se puede conseguir mediante dos vías:
- Una vía estándar para dispositivos nuevos, puestos en el mercado después de la entrada en vigor del MDR.
- Otra vía especial para dispositivos puestos en el mercado durante la vigencia del MDD, antes de que el MDR entrara en vigor.
En el segundo caso, el Artículo 120 del MDR establece una vía para que los dispositivos puestos en el mercado y certificados bajo MDD se puedan seguir comercializando en base a su certificación existente.
Sin embargo, para que un fabricante pueda declarar cumplimiento del MDR a través de su certificado MDD en virtud del Artículo 120 del MDR, se deben cumplir tres condiciones:
- Tener implantado un Sistema de Gestión de la Calidad antes del 26 de mayo de 2024, de acuerdo a la norma ISO 13485. No es necesario que esté certificado, pero debe estar implantado con anterioridad a esa fecha. En el caso de AI Labs Group SL, esto se puede evidenciar en el
Anexo III: Certificado de mantener un Sistema de Gestión de la Calidad de acuerdo a la norma ISO 13485y elAnexo VII: Declaración de la dirección sobre el Sistema de Gestión de la Calidad. - Tener un contrato vigente con un Organismo Notificado, por el que se pueda evidenciar que se presentó una solicitud de transición al MDR al organismo notificado antes del 26 de mayo de 2024. En el caso de AI Labs Group SL, esto se puede evidenciar en el
Anexo IV: Contrato con un Organismo Notificado con fecha previa al 26 de mayo de 2024y elAnexo VI: Carta del Organismo Notificado confirmando el estado de solicitud formal en relación a la disposición transitoria del MDR. - Que el dispositivo sanitario no haya experimentado cambios significativos. En el caso de AI Labs Group SL, esto se puede evidenciar en el
Anexo V: Declaración de Conformidad con MDD, incluyendo aviso de que el dispositivo no ha experimentado cambios significativos.
Si se cumplen estas condiciones, el fabricante de un dispositivo sanitario certificado bajo MDD puede comercializar su dispositivo sanitario dando cumplimiento a MDR.
AI Labs Group SL puso su dispositivo sanitario en el mercado antes de la entrada en vigor del MDR, como se puede observar el Anexo I y el Anexo II, por lo que la vía de cumplimiento del MDR adecuada es la dispuesta en el Artículo 120 del MDR.
Inicialmente, el MDR dio de plazo hasta el 26 de mayo de 2024 para que los dispositivos certificados bajo MDD se certificaran también en MDR. Sin embargo, la Comisión Europea después publicó el Reglamento 2023/607, que modifica los plazos transitorios del MDR, ampliando el plazo hasta el 31 de diciembre de 2028.
Esto significa que durante todo el periodo de ejecución de la licitación, que termina antes del 31 de diciembre de 2028, AQuAS podrá utilizar sin ningún riesgo el producto certificado bajo MDD, siempre que cumpla con las condiciones del Artículo 120 MDR. Después de ese plazo, el fabricante deberá haber completado el proceso de certificación bajo MDR; sin embargo, esto no es relevante para la presente licitación, debido a los plazos ya mencionados.
Por otro lado, cabe mencionar que la clasificación del producto sanitario que solicita la licitación es diferente bajo el MDD y el MDR. El mismo producto, con la misma finalidad prevista, es un dispositivo Clase 1 bajo MDD, y un dispositivo sanitario Clase II bajo MDR. Sin embargo, desde el punto de vista regulatorio y para la presente licitación, esta diferencia de clasificación es irrelevante dado que la vía de cumplimiento se basa en la disposición transitoria del Artículo 120 MDR, que permite la comercialización del dispositivo bajo su certificación MDD.
Exclusión no justificada
Discriminar a fabricantes que pusieran en el mercado su dispositivo sanitario con anterioridad al periodo de vigencia del MDR, a pesar de que cumplan actualmente con las condiciones establecidas en el artículo 120 del MDR, podría suponer una limitación indebida de la competencia, contraria a los principios de igualdad, libre concurrencia y no discriminación establecidos tanto en la normativa de contratación pública como en el Derecho europeo.
A este respecto, en fecha 08/08/2025 09:04, la Agència de Qualitat i Avaluació Sanitàries de Catalunya publicó una respuesta a una pregunta que decía:
Solicitud de aclaración sobre dispositivos sanitarios que cumplan con el MDR en base al Artículo 120 (...) ¿podrían justificar técnicamente por qué se excluyen dispositivos legalmente conformes con el régimen transitorio establecido por el MDR?
La respuesta de AQuAS decía:
un dispositivo médico certificado según el MDR cumple todos los requisitos europeos actuales (y más estrictos) de seguridad, eficacia y trazabilidad (especialmente en aspectos como trazabilidad, figura responsable del cumplimiento normativo, requerimientos de evidencia clínica o información y transparencia); mientras que un dispositivo certificado bajo la MDD pero en transición conforme al artículo 120 de la MDR está sujeto a sólo a ciertas obligaciones del MDR y solo hasta el final del periodo transitorio.
Esta respuesta es parcialmente correcta, pero omite una pieza de información crucial. Cuando un dispositivo certificado bajo MDD sigue la vía del artículo 120 del MDR para dar cumplimiento al MDR, automáticamente le aplican los requisitos de seguridad, eficacia y trazabilidad del MDR.
Es decir, no es cierto que un dispositivo certificado bajo MDD en transición conforme al artículo 120 del MDR esté sujeto a menos requisitos, ya que el artículo 120 establece que el fabricante debe cumplir con los requisitos del MDR, incluyendo la vigilancia del mercado y la trazabilidad. Por lo tanto, el dispositivo de AI Labs Group SL cumple exactamente los mismos requisitos que un dispositivo certificado directamente bajo MDR.
Y en el caso concreto de AI Labs Group SL, además, el organismo notificado BSI ya ha certificado que el proceso de fabricación y el sistema de gestión de calidad cumplen con los requisitos del MDR, como se puede ver en el Anexo III: Certificado de mantener un Sistema de Gestión de la Calidad de acuerdo a la norma ISO 13485. Es decir, que no sólo AI Labs Group SL está igualmente obligada a cumplir con los requisitos del MDR que cualquier otro fabricante, sino que además su SGC ha sido certificado por un organismo notificado.
Respetuosamente, la respuesta de AQuAS no es correcta, ya que un dispositivo certificado bajo MDD en transición conforme al artículo 120 del MDR sí cumple con los requisitos del MDR, incluyendo seguridad, eficacia y trazabilidad, y por tanto esta justificación no excluye que haya una exclusión no justificada contra dispositivos certificados bajo MDD antes de la entrada en vigor del MDR.
Anexos
| Anexo | Descripción |
|---|---|
| I | Registro de Dispositivo Sanitario en EUDAMED |
| II | Licencia de fabricación de producto sanitario emitida por la AEMPS, que incluye la relación de productos sanitarios de la Entidad Colaboradora. |
| III | Certificado de mantener un Sistema de Gestión de la Calidad de acuerdo a la norma ISO 13485 |
| IV | Contrato con un Organismo Notificado con fecha previa al 26 de mayo de 2024 |
| V | Declaración de Conformidad con MDD, incluyendo aviso de que el dispositivo no ha experimentado cambios significativos |
| VI | Carta del Organismo Notificado confirmando el estado de solicitud formal en relación a la disposición transitoria del MDR |
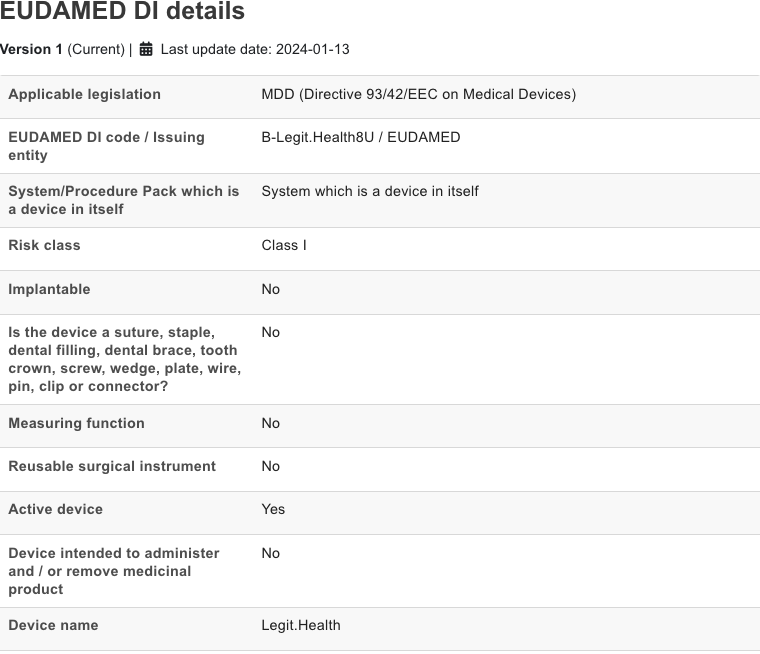
Anexo I: Registro de Dispositivo Sanitario en EUDAMED
EUDAMED es el registro público de la Comisión Europea en el que se registran los dispositivos sanitarios, de acuerdo al artículo 33 del Reglamento (EU) 2017/745 (MDR). Esta información está disponible al público a través del enlace: https://ec.europa.eu/tools/eudamed/

Anexo II: Licencia de fabricación de producto sanitario emitida por la AEMPS



Anexo III: Certificado de mantener un Sistema de Gestión de la Calidad de acuerdo a la norma ISO 13485

Anexo IV: Contrato con un Organismo Notificado con fecha previa al 26 de mayo de 2024

Se adjunta sólo la página de firma, por ser ésta la que contiene la fecha.
Anexo V: Declaración de Conformidad con MDD, incluyendo aviso de que el dispositivo no ha experimentado cambios significativos

Obsérvese que la versión del dispositivo es 2.1, por eso la fecha es 2023. Sin embargo, para la versión 1.0, la fecha es de 12 de octubre de 2020.
Anexo VI: Carta del Organismo Notificado confirmando el estado de solicitud formal en relación a la disposición transitoria del MDR

Declaración de Conformidad y marcado CE
A continuación se incluye una muestra de la Declaración de Conformidad del fabricante de acuerdo al MDR. El símbolo marcado "CE 2797" con el identificador BSI se aplicará al etiquetado y documentación, acompañando el número de Marcado CE MDR UE 2017/745.
| Identificación única del dispositivo | (01)8437025550005(10)1.1.0.0(11)YYYYMMDD | |
| Versión | (10) 1.1.0.0 | |
| Fabricante | Legit.Health Legit.Health (AI Labs Group, SL) BAT Tower, Gran Via 1, 48001, Bilbao, Vizcaya (España) |
| Marcado CE MDR UE 2017/745 | |
| Dispositivo médico | |
| Consulte las instrucciones de uso electrónicas | |
| Precaución |
Este etiquetado es un borrador de lo que ya está siendo auditado por el organismo notificado BSI, quien ya ha incluso realizado su revisión y ya ha certificado el sistema de gestión de calidad. Sin embargo, aún no ha emitido el certificado final, motivo por el cual AI Labs Group SL se acoge a la disposición transitoria del MDR (artículo 120) para poder satisfacer los requisitos de la licitación AQUAS-2025-42.